Planning Sample Sizes for Clinical Studies of Medical Devices and IVDs: The 6 Questions Your Statistician Must Answer
If you have to conduct a clinical investigation for your medical device or clinical performance study for your IVD, you won’t be able to avoid sample size planning. And you will need to avoid the following two problems when calculating the sample size:
- Problem 1: You select too big a sample size, which makes things more difficult than is necessary, wasting time, money and resources, and delaying the authorization process.
- Problem 2: Your sample size is too low. In this case, you will probably be unable to demonstrate the intended effect of your device and/or the notified body will not recognize the study due to a lack of data reliability and robustness.
Therefore, you need to accurately identify the exact sample size needed so that you don’t spend additional time and effort on your study or collect unreliable data and run the risk of having to repeat the study or even of endangering patients as a result.
This article introduces you to the 6 questions you need to answer in order to calculate the right sample size.
Note on authorship: Dr. Thomas Keller of ACOMED statistik is the co-author of this article. He helps Johner Institute clients calculate sample sizes for clinical investigations of medical devices and performance studies of in vitro diagnostic medical devices (IVDs).
1. Why you have to plan sample sizes for clinical studies
The general safety and performance requirements for medical devices and in vitro diagnostic medical devices (IVDs) are established in Annex I of the MDR and Annex I of the IVDR, respectively. Manufacturers demonstrate compliance with these requirements by testing, verifying and validating the device concerned and its components. This evidence also includes the clinical evaluation for medical devices – with regard to IVDs, the IVDR refers to performance evaluations.
When manufacturers are planning an experiment to collect valid data for their device, the question of ‘how many samples, subjects or data sets do we need to investigate?’ always comes up.
Sometimes, standards and guidance documents help calculate the sample size for a study or for individual experiments (e.g., the FDA guidance documents or, currently for SARS-CoV-2 tests, the WHO instructions). However, such guidance is not available for all devices and tests. In the absence of concrete specifications, manufacturers must calculate the sample size for the planned study and the study objective themselves.
Good study planning will enable you, as a manufacturer, to both reduce the sample size to the necessary level and collect reliable, robust and high-quality data.
Additional information
Read more on clinical investigations of medical devices here (German) and performance evaluations of IVDs here.
2. The regulatory requirements for statistics in clinical studies
EU Regulation 2017/745 on Medical Devices (MDR) and EU-Regulation 2017/746 on In Vitro Diagnostic Medical Devices (IVDR) require, in the definitions in their respective Article 2, clinical investigation plans for medical devices and performance study plans for IVDs to describe “statistical considerations”.
Definition: Performance study plan
“means a document that describes the rationale, objectives, design methodology, monitoring, statistical considerations, organization and conduct of a performance study”
Source: IVDR, Article 2(43)
The IVDR also states that a performance study for IVDs is a “study undertaken to establish or confirm the analytical or clinical performance of a device” (IVDR, Article 2(42)). Therefore, in the case of IVDs, the required “statistical considerations” affect both experiments to demonstrate analytical performance parameters and clinical performance studies.
Definition: clinical investigation plan
“means a document that describes the rationale, objectives, design, methodology, monitoring, statistical considerations, organization and conduct of a clinical investigation”
Source: MDR, Article 2(47)
a) IVD performance evaluations: IVDR
The IVDR requires IVD manufacturers to look, at an early stage, at the statistical methods they intend to use for the performance evaluation. As early as the performance evaluation plan, “appropriate statistical tools, used for the examination of the analytical and clinical performance of the device” have to be described (see IVDR, Annex XIII, Section 1.1).
Manufacturers usually document the detailed procedure for demonstrating the analytical or clinical performance of an IVD in separate plans.
However, IVD manufacturers will be searching in vain if they look in the IVDR for guidance on how to prepare a plan for the analytical performance evaluation – the IVDR merely lists the performance parameters to be tested in Annex I, Section 9.1.a). In contrast, in Annex XIII, Section 2.3.2., the IVDR details the requirements for the content of a clinical performance study plan. In Section 2.3.2, subsection j), it specifies that, as an IVD manufacturer, you must:
- Justify the design of the clinical performance study
- Demonstrate the scientific robustness and validity of the study
- Describe the statistical design and measures taken to minimize bias
b) Clinical investigation of medical devices: MDR
The requirements for clinical investigations of medical devices are described in Annex XV of the MDR. Section 2.1 states that “the clinical investigations shall include an adequate number of observations to guarantee the scientific validity of the conclusions.”
In Annex XV, Chapter II, Section 3, the MDR establishes the required contents of a clinical investigation plan. Subsection 3.6 addresses the design of the clinical investigation and the “evidence of its scientific robustness and validity” and specifies the parameters and variables to be taken into account. Section 3.7 makes it clear that manufacturers must justify the statistical considerations and conduct a power calculation for the sample size, if this is applicable to the clinical investigation in question.
c) Planning clinical investigations according to the state of the art
ISO 14155:2020
The standard ISO 14155:2020 “Clinical investigation of medical devices for human subjects – Good clinical practice” describes good clinical practice for clinical investigations of medical devices in humans. Annex A of the standard specifies the information that a clinical investigation plan must contain. More detailed guidance on statistical design, including the sample size calculation, can be found in Annex A.7 of ISO 14155:2020. The standard is referenced in the MDR in recital 64. However, the MDR refers to the ISO 14155:2011 version. The draft of the standardization request also refers to the 2011 version. In the older version, however, the section on statistical considerations is much shorter. Therefore, we recommend using the current version, ISO 14155:2020.
ISO 20916:2019
More specific guidance on good study practice for clinical performance studies of IVDs can be found in standard ISO 20916:2019 “In vitro diagnostic medical devices – Clinical performance studies using specimens from human subjects – Good study practice”. The IVDR references the standard published in 2019 in recital 66. The standard provides a kind of standard operating procedure for the planning and conduct of a clinical performance study. In section 5.3, the standard emphasizes that the design of a performance study depends on the sample size of calculation and the planned statistical analysis. In addition to the IVDR, the standard specifies the content of the performance study plan. These specifications partly overlap with the requirements of the IVDR, but also contain some additional information and more specific requirements. Thus, the standard names the parameters and variables that play a role in the collection of sample sizes. We discuss these factors in more detail in the next section.
STARD 2015
The “Reporting Guidelines” according to STARD 2015 also provide important information for manufacturers to consider when designing a diagnostic performance study. Although the guideline describes how to report the results of diagnostic studies, it is also a useful tool for study planning.
CLSI guidelines
IVD manufacturers can also find valuable guidance and specific instructions on how to plan analytical performance and clinical performance experiments in the Clinical and Laboratory Standards Institute (CLSI) guidelines. The CLSI guidelines represent the state of the art for IVD performance evaluations. EU Directive 98/79/EC on in vitro diagnostic medical devices (IVDs) previously referenced the CLSI guidelines via the harmonized standard EN ISO 18113-1:2011.
d) Requirements from the perspective of notified bodies and authorities – authorization of a clinical investigation or a performance study
For IVDs, Article 58 of the IVDR requires an application for authorization of the performance study to be submitted before certain performance studies can be conducted. In Article 67, the IVDR describes the criteria that the people reviewing the application for authorization must follow. This includes but is not limited to an assessment of the reliability and robustness of the data generated by the performance study. The reviewer should also consider the following, among other things:
- The statistical approach
- The study design
- Methodological aspects
- The sample size
- The selected comparators
- The choice of endpoints
An application for authorization must be submitted for clinical investigations of medical devices. For devices with a low safety risk, manufacturers may be exempted from the requirement to obtain authorization (see the Verordnung über klinische Prüfungen von Medizinprodukten (MPKPV)). To assess an application for authorization of a clinical investigation, reviewers should, according to Article 71 of the MDR, assess the reliability and robustness of the data generated in the clinical investigation, among other factors. The assessment criteria given in the MDR are the same as the aspects previously listed above.
Conclusion: The competent authority cannot authorize a clinical study if no systematic study and sample size planning has been carried out.
3. Sample size planning
When planning a performance study for IVDs or a clinical investigation for medical devices, manufacturers often ask how many patients, samples or data sets are needed for the study. But instead of an answer, they often initially only get counter-questions from the statisticians they ask for advice.
That's because, in order for statisticians to be able to calculate the sample size, they must already have the results of the study. However, manufacturers want to plan how to obtain these results. This is the sample size paradox.
Summary
In this section, you will learn the six questions you need to ask in order to be able to calculate sample sizes for clinical studies. You should get help from experienced statisticians when trying to answer the questions so you can plan a valid sample size quickly and without friction losses.
In the case of a complex study designs, there are additional technical issues, and these are also mentioned in this article.
a) 6 questions for calculating sample sizes in clinical studies
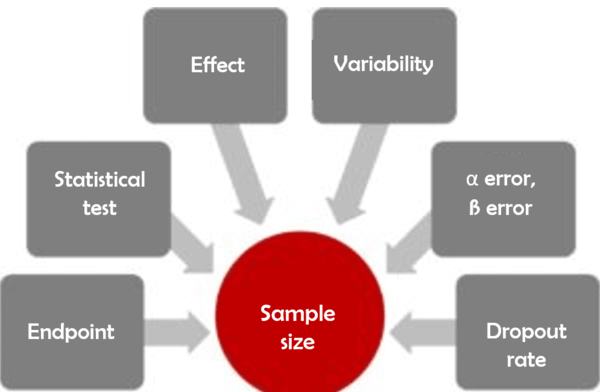
The endpoints of an IVD performance study could be, for example, diagnostic sensitivity and specificity. For a medical device for wound care, the endpoint could be, for example, the time to wound healing. Statistical tests are methods used to validate the data collected through the study. The beta error describes the risk for the manufacturer, whereas the alpha error represents the risk for the general public. The dropout rate estimates how many subjects/samples will not be evaluable.
1st question: what endpoint should be used for the study objective?
The endpoint is a statistical measure used to measure whether the study objective has been achieved or not. The selected endpoints can vary depending on the device being evaluated and the nature of the study.
For both analytical and clinical performance evaluations of IVDs, endpoints are to some extent dictated by the general safety and performance requirements established in Annex I, Section 9.1 of the IVDR. When demonstrating the clinical performance of an IVD, the endpoint is usually a proportion. This can be the proportion of true positive test results, i.e., the diagnostic sensitivity. The proportion of true negative test results represents the diagnostic specificity. The endpoint of an experiment to demonstrate the repeatability could, for example, be the coefficient of variation for tests performed repeatedly under the same conditions.
For medical devices, endpoints that describe the efficacy and benefit of the device on the one hand and, on the other, its safety must be selected. The endpoint of a study intended to demonstrate the safety of a medical device could, for example, be the proportion of patients with a complication as a result of using the device.
The evaluation of a medical device for wound healing may, for example, have the following endpoints:
- A quantitative variable, e.g.,
- the area of the wound at a certain point in time or
- the time to healing
- The proportion of patients with a reduction in wound area of at least 50% at a defined time point
- A patient-reported outcome (PRO) on subjectively perceived health status – e.g., based on a score that assesses pain, cosmetic outcome or effect on quality-of-life
- A surrogate parameter (German), such as the presence of certain cells or biomarkers in the wound
As the example shows, numerous factors usually have to be taken into account when selecting endpoints for clinical investigations of medical devices.
2nd question: which statistical test should be used as proof?
A statistical test should show that the study data collected support the proposition to be demonstrated (experimental hypothesis) (e.g., test A has a higher diagnostic sensitivity than test B).
The statistical test used depends, firstly, on the previously selected endpoints and, secondly, on the design of the study. They can be statistical tests that demonstrate a difference in means, tests to compare two proportions, tests to demonstrate non-inferiority, among many others. However, values can also simply be measured – in statistics this is known as estimation. Manufacturers must present these values along with the uncertainty. The confidence interval is usually used for this.
For antigen tests for the detection of the SARS-CoV-2 coronavirus, the WHO has published guidelines that specify, for example, the statistical procedure for detection as well as the bounds (see below). According to these guidelines, the lower limit of the confidence interval should ideally be equal to or greater than the target value.
If you need support selecting a suitable statistical test or answering any other question given here, please feel free to contact us via our contact form.
3rd question: what effect is expected?
Describing the expected effect and its variability (see the next point) is certainly the most difficult part. The paradox mentioned above once again becomes clear:
the outcome of the study has to be defined during the planning of a study.
To do this, manufacturers have to make assumptions during the planning stage about what quantitative effect they expect as the outcome of the investigation or study. For example, depending on the device, you can use various different methods to determine the achievable diagnostic quality, the expected mean difference or the complication rate:
- Research the literature and use the results from comparable studies (systematic literature search)
- Work out the requirements of the market or the competition (state of the art)
- Determine the minimum difference of interest or minimum relevant difference in the medical context (state of the art) For example, for patient-reported outcome (PRO) questionnaires, the minimum difference of interest is a property to be specified and validated by the person creating the questionnaire.
- Where they are available, use the specifications found in guidelines: For antigen tests for the detection of the SARS-CoV-2 pathogen to be used in suspected cases of COVID-19, the WHO guidelines consider a diagnostic sensitivity/specificity of ? 80%/? 97% to be acceptable. The desired target values are ? 90%/? 99%.
4th question: what is the variability of the expected effect?
To specify the expected variability (standard deviation) of the data within the study population, manufacturers can use information from similar studies in the literature as well as statistical estimates.
Manufacturers often obtain data during internal preliminary tests that they can use to derive conclusions regarding the standard deviation. As an initial approximation, you can calculate the standard deviation, for example, from the underlying measurement range as a quarter or a sixth of the range.
With proportions, the variability is already given by the proportion itself. For these endpoints (e.g., complication rates, measures of diagnostic quality), the information on variability is already implicitly available.
5th question: what is the size of the alpha error and the beta error?
The alpha and beta errors are also known as type 1 and type 2 errors. They give the probability of a false positive or a false negative test result. You can also interpret the two errors as risks.
- The alpha error represents the risk for the general public that an effect is shown in the study when in fact this effect does not exist. In other words, that the study results would show a better device effect or performance than is actually the case.
- The beta error, on the other hand, describes the manufacturer's risk that the study will fail to detect an effect that does exist. In this case, the results of the study would make the device look worse than it actually is.
Both these errors have “standard” values: The beta error, as the “manufacturer's risk” is typically between 10% and 20%. The alpha error, i.e., the risk for the general public, should be much lower, e.g., 5%. For example, Annex A.7 of ISO 14155:2020 states that for clinical investigations, alpha errors of 5% for two-sided testing do not require further justification.
6th question: what is the study's expected dropout rate?
The sixth question you should ask when planning the sample size for a clinical study looks at the expected dropout rate. You have to consider whether you are expecting to lose subjects or test results during the study.
These dropouts are quantified and included in the sample size calculation to make sure that the calculated sample size provides reliable and robust study results.
b) Additional technical issues in the case of complex study designs
Depending on the underlying study design and the objective of the study, you may have to consider other aspects when planning the sample size.
These aspects include, for example, the allocation ratio. This describes the ratio at which patients or samples are assigned to each study group. A 1:1 ratio is generally optimal but other ratios can also be justified. This has to be decided based on the planned study.
If several endpoints are being investigated together or if more than two groups are being compared, the associated multiple testing increases the sample size. In this context, multiple testing means the simultaneous evaluation of several endpoints, where at least one endpoint must be demonstrated. In these cases, the value for the alpha error (see section 3a) question 5) must be reduced for it to still be acceptable. The easiest, but also most conservative, way of achieving this is to divide the alpha error by the number of comparisons (Bonferroni correction).
Multiple testing is also relevant if you want to conduct an interim analysis during the study. Since multiple testing can have the effect of increasing the sample size, you should carefully consider beforehand whether an interim analysis is required and what the consequences of such an analysis would be.
4. Examples of sample size calculations
Once you have worked through the above sample size planning considerations for the planned study, you are ready to calculate the sample size. A computer can generally do this in a matter of seconds. However, the result of this calculation is usually not a single sample size, but different scenarios. The scenarios give sample sizes as a function of varying factors. Together with the statistician, you can evaluate the different scenarios by balancing the resulting uncertainty of a scenario against the feasibility of such a study.
a) Example 1: Sample size planning for IVD clinical performance studies
The following example illustrates the iterative approach to sample size planning for an IVD clinical performance study:
The IVD being investigated is a test for the diagnosis of a disease with a low prevalence, e.g., a screening test for cancer. The clinical study should provide the most unbiased evidence possible that the test is suitable for its intended purpose of screening the entire population above a certain age. Care must be taken to ensure that the study population represents the same clinical spectrum as the population in which the test is to be used. There must not be a spectrum bias (e.g., only severe cases). The inclusion criterion is the intended use of the test, not the known disease status of the patient.
The objective of the study is to demonstrate that the diagnostic sensitivity of the test is above a certain predefined limit, e.g., 75%. For diagnostic specificity, 90% is required. These minimum requirements are derived from, e.g., the medical state of the start for the example screening test. The manufacturer of the IVD also expects its IVD to have a diagnostic quality of 85% sensitivity and 95% specificity.
Based on the low prevalence in a screening test (generally ? 10%), the acceptance criterion for the diagnostic sensitivity (in this case, 85%) generally determines the required sample size for the study. After answering the six questions for calculating the sample size, manufacturers, if necessary together with a statistician, should first determine the number of ill patients (see Table 1, columns 1-3) because these patients form the basis for the determination of diagnostic sensitivity. The number of non-ill subjects is calculated accordingly based the number of ill people and the prevalence (see Table 1, columns 4-6).
So, what is the result of the sample size calculation for the IVD clinical performance study?
Firstly, the IVD manufacturer receives, for example, a table from the statistician listing the different scenarios. Depending on different values for the actual diagnostic sensitivity (e.g., 80%, 85%, 90%), the table shows the corresponding number of ill people (shaded in light gray). A similar calculation can be used for non-ill subjects. In this example, we assumed 92.5%, 95% and 97.5% for the actual diagnostic specificity (table, right, white background).
The example of an IVD and low prevalence shows how complex the sample size calculation and the different scenarios can become. In these cases, the number of non-ill subjects to be recruited determine the sample size (see Table 1, columns 4-6). Therefore, statisticians always give scenarios based on different prevalence assumptions.
The example shows that the sample sizes vary considerably depending on the underlying assumptions. From these scenarios, the manufacturer selects one that dictates the sample size for the IVD clinical performance study. When making this selection, the manufacturer has to balance the certainty of a successful study with reliable data against the feasibility of implementing the study in a way that ensures the study objectives are achieved. The manufacturer bases its selection on quantifiable criteria (e.g., assumptions, prevalence) and justifies it in the context of the IVD’s intended purpose.
If the manufacturer is confident that its IVD has a significantly higher diagnostic performance than that required, e.g., according to the state of the art, the study will use a smaller sample size. If the manufacturer is risk averse, it will use a lower beta error as well as more conservative assumptions for the actual performance and, therefore, a larger sample size.
1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
Beta error | Diagnostic sensitivity | Number of ill people | Number of non-ill people at a prevalence of 5% | Number on non-ill people at a prevalence of 10% | Number on non-ill people at a prevalence of 15% | Beta error | Diagnostic specificity | Number non-ill people |
20% | 80% | 563 | 10697 | 5067 | 3191 | 20% | 92.5% | 1049 |
10% | 80% | 742 | 14098 | 6678 | 4205 | 10% | 92.5% | 1371 |
20% | 85% | 133 | 2527 | 1197 | 754 | 20% | 95% | 239 |
10% | 85% | 171 | 3249 | 1539 | 969 | 10% | 95% | 301 |
20% | 90% | 54 | 1026 | 486 | 306 | 20% | 97.5% | 93 |
10% | 90% | 68 | 1292 | 612 | 386 | 10% | 97.5% | 111 |
Table 1: Table columns 1-6: Sample size calculation scenarios to demonstrate a given IVD diagnostic quality with 75% diagnostic sensitivity and 90% diagnostic specificity. Data with two different beta errors (10% and 20%) is shown.
Legend: Columns 1-3: Number of ill people with different assumptions for the true diagnostic sensitivity of the test (binomial test, 2-sided, alpha = 5%, beta = 20% or 10%);
Columns 4-6: Number of non-ill people as correctly calculated from the number of ill people (Np) and the prevalence (prev) (Np x (1-prev)/prev).
Table columns 7-9: Sample size for non-ill subjects based on the diagnostic specificity. Careful: These figures are not definitive and are for information only. For low prevalences, the sample sizes for non-ill people calculated based on the prevalence of ill people (columns 4-6) are higher and therefore definitive.
Conclusion
Sample size planning is an iterative process during which manufacturers can check the effect that changing parameters can have on the sample size. Valid acceptance criteria and defining the study objective are prerequisites for this. The sample size selection is based on quantifiable criteria for the likelihood of success and the feasibility of the study.
b) Example 2: Sample size calculation for a clinical investigation to demonstrate the safety of a medical device
For clinical investigations to demonstrate the safety of medical devices, the complication rate, for example, can be used as the endpoint. These rates usually have very low values between 0% and 5%. The associated confidence intervals are correspondingly narrow. In order to be able to make reasonably reliable predictions about the proportion of patients with complications, sample sizes are often in the three to four-digit range.
The following example is intended to illustrate the sample size calculated as a function of the endpoint – the acceptable complication rate: The expected complication rate is 2% and the manufacturer would like to demonstrate a rate of < 5% for its medical device (corresponds to the upper limit of the confidence interval (CI)). The manufacturer derives this requirement from, for example, the medical state of the art or health economics considerations (cost/benefit considerations). In this example, approximately 330 patients are required. If the complication rate were 3%, the same CI upper limit of 5% would mean that 815 patients would have to be included the study [1], [2]. If, when calculating the sample size, we also take into account that 5% of patients will not be contactable during the follow-up phase (5% dropout rate), then the sample size is increased by the factor 1/(1-0.05) = 1.053.
[1] Sample size calculation: Binomial test, 2-sided, alpha level = 5%, beta level = 20%, software PASS 20.0.
[2] One way of reducing the number of cases is to prospectively use 1-sided tests for such questions. In the example, this would result in a sample size of 253 (2%) and 631 cases (3%) respectively. However, in clinical research, alpha = 2.5% is used for 1-sided tests to prevent misuse of 1-sided tests. As a result, there is no sample size advantage. In terms of content, however, there is nothing to be said against 1-sided tests for questions where the ideal rate is 0%. If necessary, this must be discussed with the authorities in advance.
5. Sample size planning: conclusion and summary
The regulatory requirements of the IVDR and MDR do not provide any specific guidance on the sample sizes required for clinical investigations of medical devices and IVD performance studies. Instead, the manufacturer can and must define and justify the sample size of a study on a device-specific basis based on the study objective.
Early involvement of a statistician in study planning is helpful for estimating the sample size and the planned statistical analysis. It can help manufacturers ensure the scientific robustness and validity of the data generated and avoid unnecessarily high costs for clinical investigations or performance studies.
The sample size calculation is a kind of “negotiation” within valid acceptance limits. It usually provides different scenarios rather than a single sample size. Together with the manufacturer, statisticians evaluate these scenarios with regard to the uncertainty and feasibility of a study.
Sample size calculation support
The Johner Institute's team will be happy to help you:
- Create the clinical evaluation for your medical device
- Plan the clinical investigation of your medical device
- Define your strategy for evaluating the performance of your IVDs
- Plan the clinical performance study for your IVD
- Calculate the sample size for your clinical investigation or clinical performance study

Author:
Dr. Catharina Bertram

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.