Regulatory Update or How to Stay Up to Date with Regulatory Requirements
Tuesday, April 28, 2020
Medical device manufacturers are required to carry out a regular “regulatory update.” Because as strange as it sounds, regulatory requirements oblige manufacturers to continuously monitor and evaluate changes to regulatory requirements and take any necessary measures.
Keeping track of thousands of regulatory requirements is a challenge. Manufacturers should have a clear understanding of the typical mistakes that they should avoid at all costs in order to, firstly, have certainty during audits and, secondly, to save themselves unnecessary time and effort on their “regulatory update.”
1. Regulations that a regulatory update must observe
The regulatory map is as complex as it is extensive:
- National laws, e.g., MPG (German Medical Devices Act), MPDG (Medical Devices Implementation Act), Swiss Therapeutic Products Act (Heilmittelgesetz), US Food, Drug, and Cosmetic Act
- National ordinances, e.g., German Safety Plan for Medical Devices (MPSV), German Medical Device Operating Ordinance (MPBetreibV)
- Other national publications, e.g., the German National Working Group (NAKI)
- EU regulations, e.g., MDR and IVDR
- EU directives, e.g., MDD, IVDD and AIMDD
- EU guidance, e.g., the MDCG documents
- Hundreds of national and international standards, e.g., ISO 13485 and ISO 14971
- Common specifications
- Well over 600 FDA guidance documents, e.g., on cybersecurity and vigilance
- Implementation guidelines, e.g., on IT security and on machine learning
- Publications from organizations such as the IMDRF and NBMED
Further Information
A further overview of regulations with the corresponding links can on the “Regulatory Affairs” page of our website.
2. Regulatory requirements for regulatory requirement research
a) EU regulations (MDR, IVDR)
Manufacturers must know and take into consideration current standards and common specifications
The EU regulations require medical devices to meet the general safety and performance requirements of Annex I (MDR Article 5(2)). These general safety and performance requirements must comply with the “generally acknowledged state of the art” (MDR, Annex I, paragraphs 1 and 4).
When demonstrating conformity, manufacturers must specify the harmonized standards, common specifications or “other solutions” applied (MDR, Annex II, Section 4(c)).
This is because Article 8 allows manufacturers to use harmonized standards to demonstrate that devices comply with the state of the art. Therefore, manufacturers must also track these standards. The MDR even explicitly states that:
changes in the harmonized standards or CS by reference to which the conformity of a device is declared shall be adequately taken into account in a timely manner.
Conclusion: medical device manufacturers must actively track standards and common specifications (CS) to make sure that they know the state of the art and can demonstrate that their devices meet it.
N.B!
As the harmonization of standards has come to a standstill, notified bodies generally expect manufacturers to follow the most recent versions of standards.
Manufacturers must describe in their QM systems how they are tracking the legal requirements
The MDR and IVDR have significantly increased the requirements for QM systems. As a result, the MDR requires:
Those procedures and techniques shall specifically cover:
— the strategy for regulatory compliance, including processes for identification of relevant legal requirements, qualification, classification, handling of equivalence, choice of and compliance with conformity assessment procedures
b) ISO 13485
Regulatory update as an explicitly required activity within the QM system
ISO 13485 also explicitly addresses the topic. It states:
“Top management shall ensure that customer requirements and applicable regulatory requirements are determined and met.”
DIN EN ISO 13485:2016, Section 5.2
Section 5.6 (“Management review”) makes clear how important this monitoring is for the standard. This management review must assess the “applicable new or revised regulatory requirements” as an input.
The output of this management review must define the “changes needed to respond to applicable new or revised regulatory requirements.”
Other device-specific regulatory requirements
In addition, ISO 13485 requires manufacturers to determine the stakeholder requirements for each device. This means, firstly, customer requirements. And secondly, regulatory requirements:
“The organization shall determine: [...] applicable regulatory requirements related to the product;”
ISO 13485 Section 7.2.1
But identifying these requirements is not enough:
“The organization shall review the requirements related to product. This review [...] shall ensure that [...] [the] applicable regulatory requirements are met; [and] the organization has the ability to meet the defined requirements.”
ISO 13485 Section 7.2.3
c) ISO 20416 (“Medical devices – Post-market surveillance for manufacturers”)
ISO 20416 considers the “regulatory update” part of the post-market surveillance. The standard explicitly states:
“Medical device organizations should monitor applicable regulatory requirements for any change to evaluate upcoming gaps, and plan for continued compliance. Standards, guidances and best practices are typically not mandatory requirements (see regulatory requirements) but describe the state of the art.
Changes in regulatory requirements, standards, guidances and best practices can suggest a change in the state of the art, impacting design and development inputs and potentially requiring design and development changes”
ISO 20416 (Draft)
d) 21 CFR part 820
The FDA does not state quite so explicitly that manufacturers have to identify the regulatory requirements and standards. But it does say that:
“Where process controls are needed they shall include: Compliance with specified reference standards or codes; “
That FDA inspectors assume that the manufacturers know and follow all the regulations should, however, go without saying.
3. Hurdles and challenges
It is common sense that manufacturers must know and comply with the regulatory requirements. But the challenges they face to do so are complex:
- Actually finding the applicable regulatory requirements
The first hurdle that manufacturers have to overcome is identifying the relevant regulatory requirements. The more countries a device is to be sold, the longer this list of regulations. - Cost of monitoring and evaluation
Once a manufacturer has found all the applicable regulations, they will be overwhelmed by the sheer volume – it’s not unusual for a manufacturer to be faced with several hundred applicable regulations.
Each document means additional time and costs for:- Monitoring
- Reading and understanding
- Recognizing differences
- Working out the necessary consequences, which can go as far as device modifications or even a “recall”
- Heterogeneity of the sources of information
But how do you stay up to date? Every source, e.g., ISO, uses different media report changes: newsletters, RSS feeds, Twitter messages, for example. Unfortunately, this information is often missing completely, meaning that manufacturers are forced to actively search for changes. - Requirements only available in the national language
But even if you find out that there are new regulations, the problem isn’t necessary solved. A lot of countries and regulatory authorities, e.g., the Chinese NMPA don’t publish some of their regulations in English or German, or only do so with a delay. - Comprehensibility
Just because a regulation is available in German or English doesn’t guarantee that you will be able to understand the requirements it establishes. Article 120(3) of the MDR is an ignominious example of a regulation that’s very difficult to understand. - Costs
Costs are another hurdle – especially for start-ups. For example, organizations such as DIN, ISO or IEC often charge several hundred euros – per standard. A total of several thousand euros each year is not unusual.
4. Typical mistakes made during the regulatory update
The Johner Institute frequently sees the following mistakes that can lead to unpleasant surprises during audits or authorization processes being made:
- The manufacturer has not identified all the requirements. In particular, national specifications and guidelines, which are not mandatory but are still required, are often missed out.
Sometimes it is even that case that not all the regulatory requirements regarding the identification of regulatory requirements have been identified.
Sometimes the company is aware of the regulations but each department keeps its own lists, and there is not a consolidated version of these lists available during the audit and management review. - The changes are identified too late or too infrequently. It is embarrassing to realize during an audit that a manufacturer doesn’t even know about an amendment to a guideline published six months ago.
- In most cases the reason for this is that there is no process and the responsibilities are not clearly defined. The regulatory affairs manager has relied on the regional office, the device manager on regulatory affairs.
- But simply knowing the changes is not enough. Often an appropriate analysis of what the changes are and an assessment of what they mean is not carried out. Such an assessment usually requires risk management, but it is often not included.
- Incomplete management review: Management only checks whether new regulations have been searched for. It does not evaluate the quality of this monitoring nor the quality of the measures resulting from new or amended regulations.
5. Regulatory update best practices
a) Define the process
Regardless of whether a process or procedure is required for the regulatory update, it is always better to have one. A corresponding standard operating procedure might include the following steps:
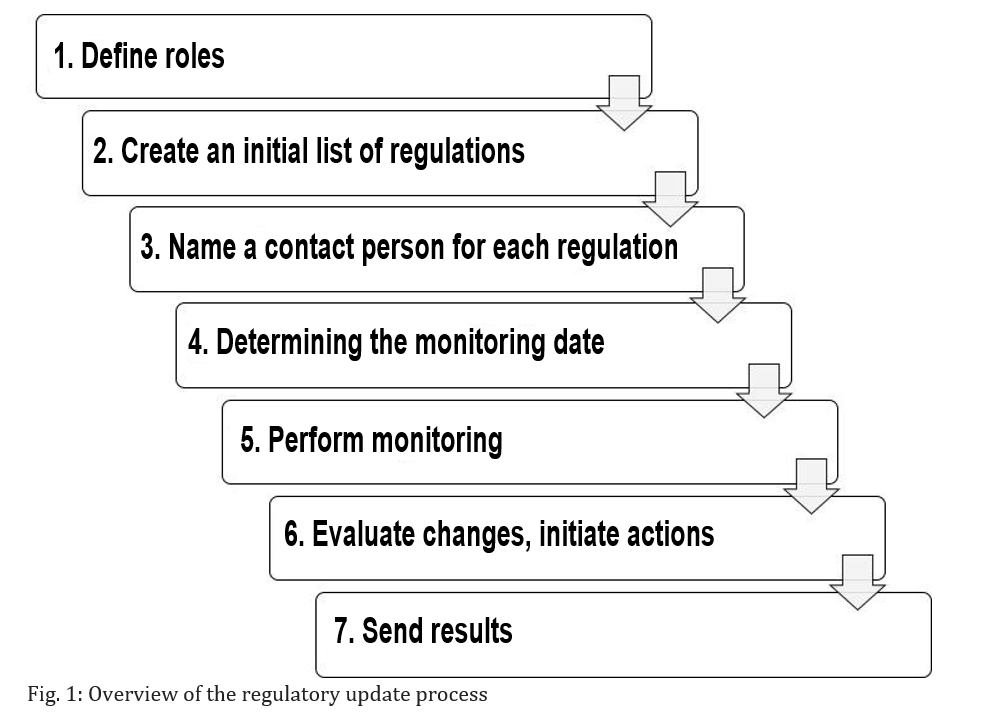
Step 1: Defining the responsible roles
Determine the roles that are responsible for creating the initial list of regulatory requirements and its continuous monitoring. Typical roles include device manager, developer, regulatory affairs and quality managers, the legal department and regional office.
Step 2: Creating an initial list of regulatory requirements
Now assign (for each device) specific people to the roles. These people will create and monitor device-specific and company-specific lists of applicable regulations. Normally, several lists are created and then consolidated.
Step 3: Naming a contact person for each regulation
Next, for each regulation, designate the person who will review changes to the regulation and act as the responsible contact person. The designated person should also document the version status in the aforementioned list.
Step 4: Determining the monitoring date
It is now necessary to define when the regulations will be checked – either for each regulation individually or for all the regulations together.
- This monitoring can be done in cycles, for example one month before the management review. An annual cycle is often not sufficient.
- The monitoring can also be linked to life cycle phases (e.g., at the start and end of development or during design reviews).
- Lastly, the monitoring can be event-based. For example, the move from the MDD to the MDR means that all the associated guidelines will have to be identified and evaluated again.
Step 5: Performing the monitoring
The selected roles or persons monitor the identified regulations in accordance with these specifications at the specified frequency and document whether there have been any changes.
Step 6: Evaluating changes and initiating possible actions
If there are any changes, the roles from step 5 inform the contact partner and request:
- An analysis of these changes
- An evaluation of the relevance of the change (use template for this and involve the risk manager if necessary)
- The initiation of the necessary actions (branching off into other processes)
- That all this is documented
Step 7: Sending the results
In each case, i.e., regardless of whether changes were identified or not, the results must be used as an input for other processes and be sent for use in these processes. This particularly includes processes such as the management review and post-market surveillance.
The “branching off into other processes” mentioned in step six can refer to, for example, the:
- Development process
- Maintenance process
- Problem solving process
- Processes for corrective and preventive actions
- Vigilance process
b) Automating the process or using automate automated processes
Sections three and four have already highlighted how challenging the regulatory update process is and described the typical mistakes made by manufacturers.
In order to avoid these errors and having to carry out repetitive activities, manufacturers should consider automating these activities. Computers can perform the process faster, with fewer errors and more cost-effectively.
For example, the Johner Institute uses crawlers to continuously monitor websites and databases and send changes to a monitoring team for analysis.
By the way, this team is responsible for monitoring regulations for a lot of manufacturers, which means that the manufacturers also benefit from economies of scale.
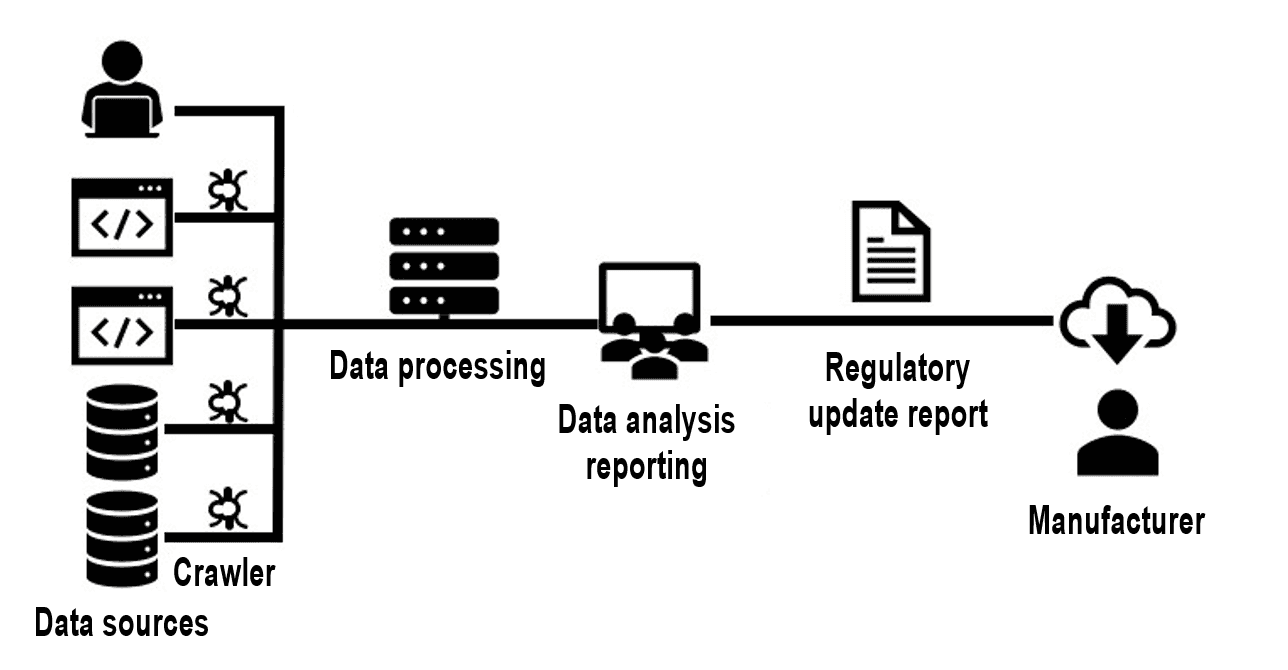
Manufacturers who do not use an automated service like the Johner Institute’s Regulatory Radar but prefer to automate the monitoring themselves should pay attention to the following:
- The more regulations there are to be monitored and the more frequent this monitoring is, the more the investment pays off.
- Complex business logic is required to avoid “false positives” and “false negatives” as much as possible. Some websites deliberately change the IDs in the HTML code on a daily basis to deter crawlers. In addition, the technical quality of some pages is questionable.
- Continuous adjustments to the software are necessary because the structure of the available information, especially on websites, changes regularly.
- The software is subject to the requirements of ISO 13485 and must be validated, and re-validated in the event of adjustments, (see Computerized Systems Validation).
6. Summary
Manufacturers must proactively monitor and evaluate changes to regulatory requirements. This is itself a regulatory requirement.
While it is true that manufacturers are not explicitly required to establish a procedure for doing this, we strongly recommend doing so. The MDR demands a “strategy” at the very least.
The growing number of regulations and the frequency with which they are updated means that manufacturers have to invest a lot of time and effort in monitoring and evaluation. Therefore, we recommend automating or outsourcing these activities as much as possible, like the pharmaceutical industry, for example, has already been doing for a while.
Anyone automating this monitoring process must validate their system.
Manufacturers must by no means limit their monitoring to regulatory requirements specific to medical devices. In fact, they must also research and comply with regulations on, for example, data protection, product safety, waste management and social law.
In addition, manufacturers should not see the regulatory update as an isolated process but as part of the post-market surveillance. This obliges manufacturers to collect, evaluate and respond to a much greater volume of information.
Further Information
Find of more on the Regulatory Radar, the Johner Institute's regulatory update service, here.

Author:
Andrea Seeck

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.