21 CFR part 820: FDA requirements for quality management systems
In 21 CFR part 820, the FDA formulates the requirements for the quality management systems of medical device manufacturers, among others. This means that the 21 CFR part 820 is a counterpart to ISO 13485.
Update: FDA is close to recognizing ISO 13485:2016 instead of 21 CFR part 820 as evidence of a quality management system.
What 21 CFR part 820 requires
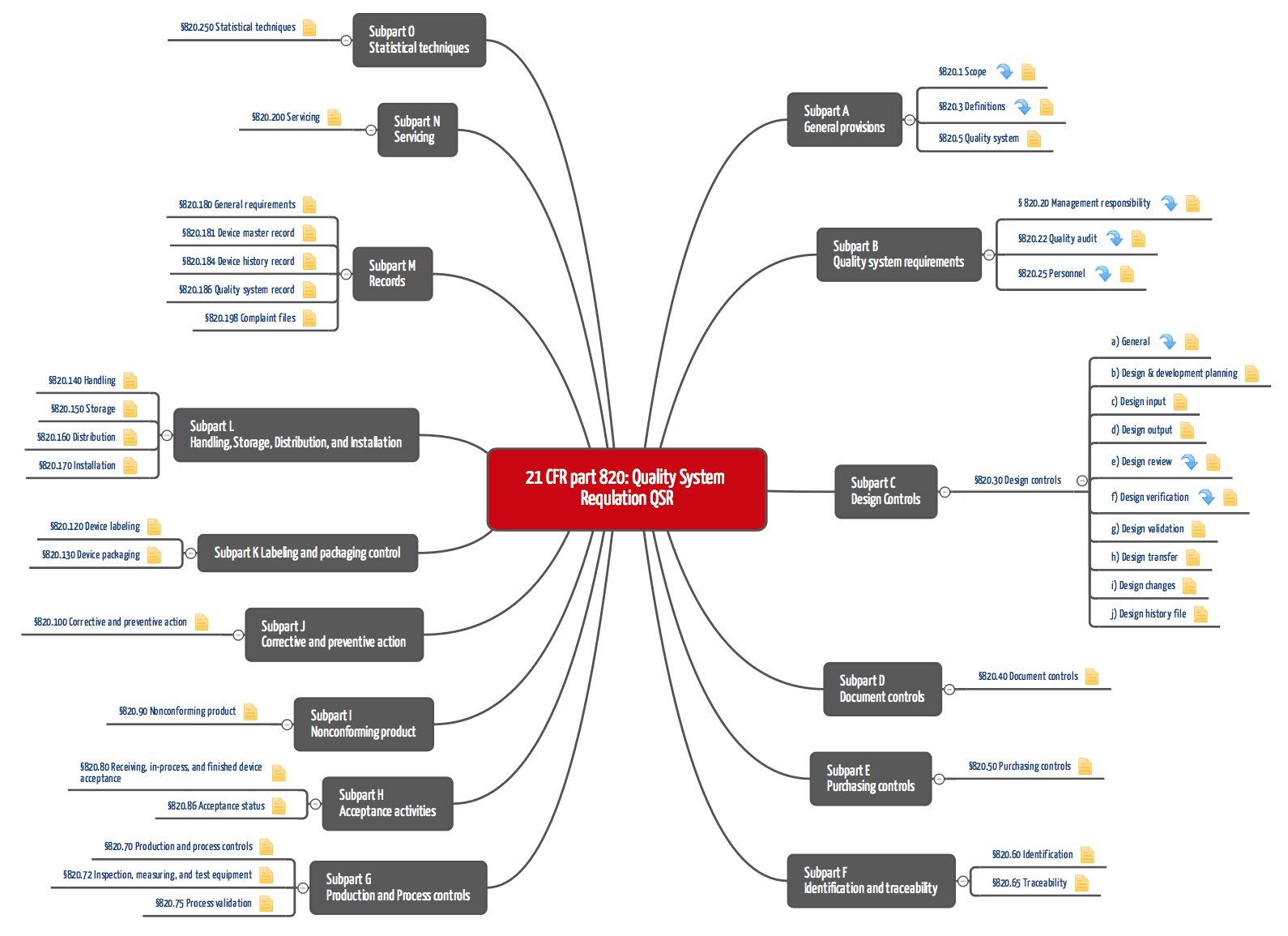
Figure 1: QSR: 21 CFR part 820 (click to enlarge)
21 CFR part 820 requires a complete quality management system, which requires that "standard" operating procedures be documented and implemented. These include:
- Document control,
- Procurement,
- Development and
- Production.
For the development department, area 820.30 with the design controls is relevant, and however, the requirements there are very general. For this reason, manufacturers of medical devices containing software, for example, are also guided by the FDA's guidance documents, which describe in more detail, for example, how the design input must be documented.
Many manufacturers underestimate the importance of the Design History File DHF (21 CFR part 820.30). This set of documents must make it possible to prove that the procedures described in 21 CFR part 820 were actually implemented - and that the documentation was not created retrospectively.
When you must comply with the Quality System Regulations (21 CFR part 820)
Depending on the medical device class, manufacturers must comply with the requirements outlined in the Food, Drug & Cosmetic Act. "General Controls "(§501 ff) and, from Class II, also comply with the "Special Controls." The "General Controls" already contain "Good manufacturing practice requirements" related to the devices' development, production, packaging, storage, and installation.
These requirements for "Current good manufacturing practice (CGMP)" are the subject of the Quality System Regulation QSR of 21 CFR part 820. Thus, these regulations must be complied with by all medical device manufacturers. However, compliance with these regulations is not checked for Class I products - unless an incident occurs.
Difference between 21 CFR 820 and ISO 13485
The extensive correspondence between the two standards is apparent. Nevertheless, there are differences between ISO 13485 and 21 CFR part 820 to be noted:
- The documentation requirements are higher, and ISO 13485 also does not recognize the logical grouping of documents into the Design History File, the Device Master Record, and the Device History Record.
- Conversely, the requirements of ISO 13485 and ISO 9001 for customer satisfaction and continuous improvement of the QM system go beyond the requirements of 21 CFR part 820.
- Furthermore, the handling of complaints and the reporting system differ significantly.
A more comprehensive comparison between the two standards is given in this document.
Tip
The differences between 21 CFR part 820 and ISO 13485:2016 are so minor that FDA is considering replacing 21 CFR part 820 with ISO 13485:2016.
At present, manufacturers can use the Medical Device Single Audit Programs MDSAP simultaneously, demonstrate compliance with the requirements of 21 CFR part 820 and ISO 13485:2016.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.