ISO 10993 and Biocompatibility - Material Certificates Are Not Enough!
Regulations such as the MDR require proof of the biocompatibility of all materials that come, directly or indirectly, into contact with patients or users.
With the right strategy, manufacturers can demonstrate compliance with the requirements of the relevant harmonized standard, ISO 10993, in a cost-effective and “audit-proof” way.
This article will give you with specific practical tips.
A test-based biocompatibility assessment provides reference data that provides a very good basis in the event of unexpected problems and that helps to quickly and directly identify unknown causes.
However, there has been a trend in the opposite direction in recent years, in other words away from testing and, therefore, away from device safety.
1. Basics
a) Risks resulting from a lack of biocompatibility
Medical device manufacturers must guarantee the safety of their devices and to minimize potential risks. These risks also include biological risks, for example those caused by:
- Chemicals that are leached from the materials in the medical device and absorbed by the body
- Implants that cause a rejection reaction
- Materials that trigger an allergic reaction
- Materials that are broken down or transformed by the body contrary to the intention of the manufacturer
- Implants and materials that do not grow into the body as desired
This article will describe factors that have a negative effect on biocompatibility.
b) Definitions of terms
ISO 10993 defines the term biological risk as follows:
Definition: Biological risk
“combination of the probability of harm to health occurring as a result of adverse reactions associated with medical device or material interactions, and the severity of that harm”
Source: ISO 10993-1
Biological safety is, therefore, the freedom from unacceptable biological risks.
The definition of the term “biocompatibility” is slightly more cryptic:
Definition: Term
“Ability of a medical device or material to perform with an appropriate host response in a specific application”
Source: ISO 10993-1
The term host response here refers to potential undesirable adverse reactions. The standard states that biocompatibility can be demonstrated through biological testing and an evaluation of the leachables.
2. Regulatory requirements for biocompatibility
a) MDR requirements
The Medical Device Regulation (MDR) puts a lot of importance on biocompatibility, as is evident even in the introductory “whereas clauses”. The corresponding requirements are explained in several places:
Annex I: General safety and performance requirements
Annex I does not explicitly mention the term biocompatibility. However, it does say:
Devices shall be designed, manufactured and packaged in such a way as to minimise the risk posed by contaminants and residues to patients, taking account of the intended purpose of the device, and to the persons involved in the transport, storage and use of the devices. Particular attention shall be paid to tissues exposed to those contaminants and residues and to the duration and frequency of exposure.
MDR, Annex I, paragraph 10.1
In the very first section of Annex I, the MDR demands that the safety and health of patients, users and third parties are not compromised and that manufacturers must eliminate or reduce risks as far as possible through safe design and manufacture”.
Annex II: Technical documentation
In Annex II, the MDR requires manufacturers to document information on all tests, including the
“test design, complete test or study protocols, methods of data analysis [...] test conclusions regarding in particular the biocompatibility of the device including the identification of all materials in direct or indirect contact with the patient or user [as well as regarding the...] physical, chemical and microbiological characterisation.”
MDR, Annex II, paragraph 6.1.b)
The MDR also contains regulations for the event that the manufacturer does not carry out any tests:
Where no new testing has been undertaken, the documentation shall incorporate a rationale for that decision. An example of such a rationale would be that biocompatibility testing on identical materials was conducted when those materials were incorporated in a previous version of the device that has been legally placed on the market or put into service;
MDR, Annex II, paragraph 6.1.b)
Annex VII: Requirements for notified bodies
This annex affects manufacturers indirectly at least, because in it the MDR requires notified bodies to:
- guarantee the competence of the notified body staff with regard to biocompatibility
- Up-to-date knowledge of the manufacturer
Notified bodies must check a manufacturer’s processes designed to ensure that the manufacturer is using the latest scientific knowledge, including with regard to the biocompatibility of materials.
b) ISO 10993-1
The standard ISO 10993-1 was already harmonized under the Medical Device Directive (MDD) and will remain so under the MDR as well (like the entire family of standards).
Therefore, most medical device manufacturers use this standard for guidance, for example when it comes to the endpoints required for the evaluation of biological safety.
The standard describes these endpoints in Table A.1 in Annex A. The following extract from the table shows some examples of the endpoints that have to be evaluated for a medical device that comes into contact with circulating blood.
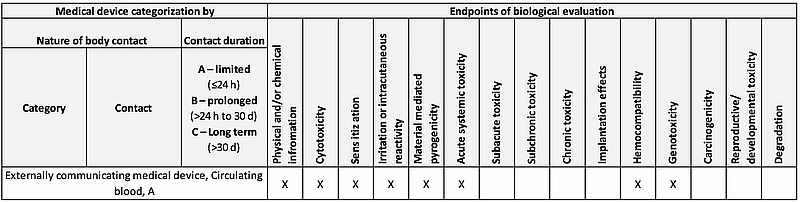
Fig. 1: Taken from ISO 10993-1 Annex A, Table A.1
N.B!
This process, which is described in the non-normative part, is not the same as the normative specifications: According to ISO 10993-1 (2018), Figure 1, and ISO 10993-2, material characterization must be carried out and animal experiments must be avoided. Material characterization also enables greater validity of the data and, therefore, higher biological safety.
The standard makes it clear that manufacturers must ensure biological safety over the entire life cycle of a medical device:
“The biological safety of a medical device shall be evaluated by the manufacturer over the whole life cycle of a medical device.”
ISO 10993-1, 4.7
This part also adds to the requirements for “freedom from unacceptable biological risks” as follows:
“The following shall be taken into account for their relevance to overall biological evaluation of the medical device: …process contaminants and residues… packaging materials… leachable substances[U11] … degradation products…”
ISO 10993-1, 4.3
3. Processing information on biocompatibility
a) Use available information
Manufacturers often gather together all the available information on the materials used to evaluate their biocompatibility, e.g.
- Material data sheets
- Certificates
- Results of research on known ingredients and additives
- Market experience
N.B!
This information is necessary and important, but it is not usually enough on its own to demonstrate biocompatibility!
b) Add missing information
The information mentioned above is a very good starting point. However, often it does not allow for complete statements on biological safety to be made. For example, there is no information that proves that there are no biological risks from
- Interactions between the various materials
- Changes in the materials during the device's life cycle
- The processing of materials during production
- The final cleaning of the device
- etc.
The list of possible influencing factors is very long. They range from hand disinfectants for employees, to impure raw materials, through to unnoticed cable wear on systems. All the factors in the following word cloud have prevented a positive assessment of biocompatibility in the past.

Fig. 2: ISO 10993 – Examples of factors that influence biocompatibility
c) Critically evaluate existing information
Manufacturers should also critically question the reliability of the available information and, for example, clarify the following aspects:
- Test methods and parameters
- Raw materials used to obtain these results
- Transferability of the results
It is good that manufacturers rely on information from competitors. However, when arguing that their products are sufficiently similar and have been used on the market for years without problems, they should consider the equivalence of all parameters such as:
- All substance and material types, such as additives and colorings, and not just the main components
- Production process and production tools
- Final cleaning: process, cleaning agent
This information is often missing and leads to gaps in arguments. Substances not listed in the data sheet are not automatically non-critical.
4. Further practical tips
The following graph reflects the Johner Institut's experience of the typical problems encountered based on our experience with several thousand medical devices.
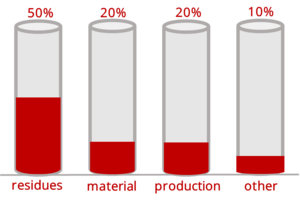
Fig. 3: Typical causes of biocompatibility problems
a) Do not rely on “established opinions”
Some manufacturers argue that cytotoxicity testing alone would be sufficient to guarantee biological safety or that devices with very short skin or mucous membrane contact should be considered non-critical. Neither of these opinions is correct and they do not comply with the MDR requirements described above.
b) Provide the necessary supporting evidence
Unfortunately, both ISO 10993 and MDR confuse us with several statements that could lead us to conclude that, for some devices, no tests are required. And in other places, there are contradictory claims. That's why we have two concrete practical tips:
- What notified bodies want to see
Notified bodies almost always want to see results of tests for the endpoints listed in the table. Or at least a reason why these tests were omitted/replaced. - Effects manufacturers must discuss
It is usually not possible to exclude in advance effects resulting from cleaning agent residues, production residues, sterilization, adhesive hardening, the evaporation of solvents, effects caused by engraving, printing, material impurities from extrusion, inadequate polymerization, packaging components, inadequate material qualities, impermissible plasticizers or other additives, coloring, etc.
c) Do not use inferior materials
A lot of problems can be avoided right from the start by using high quality raw materials. So, for example, there can be way over 200 different extractables if you use poor quality polyurethane (PUR), although not always all at once. In contrast, if you use a very good PUR, there should be no leachables.
d) Tests: make them risk-based
It takes a lot of experience to be able to set a limit above which a material can no longer be considered acceptable. And, of course, extensive knowledge of limits is essential for this.
Most medical devices do not give any abnormal results with regard to biocompatibility testing. The percentage largely depends on the material and type of device. For devices with abnormal results, however, 90% of the deviations can be argued to be acceptable straight away or after subsequent analysis, or can be rectified by simple optimization.
To better assess the risks, use your experience and knowledge of:
- The device and device type (history)
- Similar devices
- Materials
- Preparation and production of the device
5. Conclusion
Demonstrating biocompatibility cannot be done using data sheets alone. Therefore, tests are necessary.
However, these tests should not be seen as a deterrent because the costs are very manageable. That’s the experience of the biocompatibility experts at the Johner Institut who have tested several thousand medical devices.
Keeping the costs of these tests low is possible if you choose the right strategy and
- carefully research the available information
- list factors that could have an effect on biocompatibility
- draw conclusions from both of these and create chains of evidence, and
- use a risk-based approach to minimize the cost of the tests, for example, by avoiding unnecessary tests.
If you have any questions or comments, please contact our free and no obligation micro-consulting.
See other articles in Product Development

Author:
Dipl.-Ing. (FH) Sarah Gruber

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.