QMSR: The end of 21 CFR part 820?
In 21 CFR part 820, the FDA formulates the requirements for the quality management systems of medical device manufacturers, among others. Thus, 21 CFR part 820 (Quality System Regulation QSR) is or was the counterpart to ISO 13485.
1. The planned amendments to 21 CFR part 820
This is because the FDA has decided to largely "gut" 21 CFR part 820 and add a reference to ISO 13485.
- This reference is in the new section § 820.7 ("Incorporation by reference").
- Tab. 1 shows which sections have become obsolete as a result.
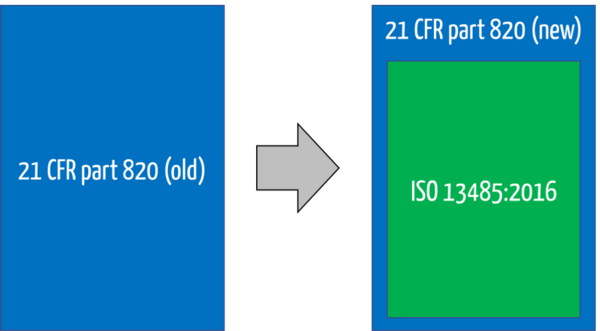
Because ISO 13485 refers to quality management systems, the FDA has decided to rename the title of 21 CFR 820. It is no longer called the Quality System Regulation (QSR) but the Quality Management System Regulation (QMSR).
Note!
With this change, the previous requirements for a design history file, a device master record, and a device history record are no longer explicitly mentioned. Nevertheless, ISO 13485 also requires similar records.
a) Comparison of the old and new 21 CFR part 820
Subparts A-D
21 CFR part 820 old | 21 CFR 820 new (QMSR) |
Subpart A – General Provisions | Subpart A – General Provisions |
| Subpart B – Supplemental Provisions |
Subpart B – Quality System Requirements | Deleted (reserved for future additions) |
Subpart C – Design Controls | Deleted (reserved for future additions) |
| § 820.35 Control of records |
Subpart D – Document Controls | Deleted (reserved for future additions) |
Tab. 1a: Comparison of the old and new 21 CFR part 820 (Subparts A-D)
Subparts E-O
21 CFR part 820 old | Planned amendments |
Subpart E – Purchasing Controls | Deleted |
Subpart F – Identification and Traceability | Deleted |
Subpart G – Production and Process Controls | Deleted |
Subpart H – Acceptance Activities | Deleted |
Subpart I – Nonconforming Product | Deleted |
Subpart J – Corrective and Preventive Action | Deleted |
Subpart K – Labeling and Packaging Control | 820.45 Device labeling and packaging controls. |
Subpart L – Handling, Storage, Distribution, and Installation | Deleted |
Subpart M – Records |
|
Subpart N – Servicing | § 820.35 Control of records (only regarding Servicing Records) |
Subpart O – Statistical Techniques | Deleted |
Tab. 1b: Comparison of the old and new 21 CFR part 820 (Subparts E-O)
b) Differences compared to ISO 13485
The Quality Management System Regulation in part 820 and ISO 13485 are not completely congruent:
Aspect | Differences |
Scope | The scope (§ 820.1) differs overall. For class I devices (excluding, for example, devices containing software), the FDA waives the requirements of Chapter 7.3 of ISO 13485 ("Design and Development"). |
Definitions | The FDA adds its own definitions to ISO 13485, such as "Component," "Customer," "Design Validation," and "Process Validation." The terms "Manufacturer" and "Product" are defined differently. |
Product identification, traceability and labeling | Here, the FDA is more specific and requires the UDI as described in § 830 and "traceability" according to § 821. The labeling requirements are also more specific than those of ISO 13485, just as in the MDR, and are found in the new § 820.45. |
Vigilance and communication with authorities | For the more specific requirements, FDA refers to Parts § 803 and § 806. |
Documentation | The FDA specifies in more detail the content of records of customer complaints and service activities. |
Tab. 2: Differences between 21 CFR part 820 (new) and ISO 13485
Further information
You can find the rationale and detailed description of the changes on the FDA website. You will need to scroll way down or search the website for the term "4. Revise part 820 to read as follows:"
An overview of all FDA requirements can be found here under the keyword FDA.
c) Timing and transition periods
The final rule for the QMSR was published on February 2, 2024. It will become valid and therefore applicable with a deadline of 2 years, i.e., February 2, 2026. Until then, the old QSR must still be followed.
2. 21 CFR part 820 before referencing ISO 13485
a) Overview
The Quality System Regulations consist (before referencing) of Subparts A through O, which include Sections 1 through 250 (see Tab. 1 and Fig. 2).
Part 820 requires a complete quality management system, which requires that the “standard” operating procedures must be documented and implemented. These include:
- Document control
- Purchasing
- Development
- Production
b) Applicability of 21 CFR part 820
Depending on the class of the medical device, the manufacturers must comply with the General Controls (§ 501 ff) laid down in the "Food, Drug & Cosmetic Act" and, from class II, also with the "Special Controls." The "General Controls" already include "Good manufacturing practice requirements" that relate to the design, production, packaging, storage, and installation of the devices.
It is precisely these requirements for "Current good manufacturing practice (CGMP)" that are the subject of the Quality System Regulation QSR. Thus, these regulations must be complied with by all medical device manufacturers and also other players such as contract manufacturers. Only a few class I devices are exempt or GMP-exempt. The FDA reviews compliance with CGMP by inspection.
c) Development requirements
Area 820.30, with the design controls, is particularly relevant for the development department. This even applies to the development of class I medical devices if they contain software or are software.
The requirements of 820.30 are very general. For this reason, manufacturers of medical devices that contain software, for example, are also guided by the FDA's guidance documents. These describe in detail how, for example, the design input must be documented.
Many manufacturers underestimate the importance of the Design History File DHF (820.30 j). This set of documents must make it possible to prove that the procedures described in 21 CFR part 820 were actually implemented and that the documentation was not created retrospectively.
d) Difference between 21 CFR 820 and ISO 13485
AnsonGroup has published a comparison of the requirements of ISO 13485 and FDA QSR. The extensive correspondence between the two standards is obvious. Nevertheless, there are differences between ISO 13485 and part 820 to consider:
- The requirements for the documentation are higher. Also, the logical grouping of the documents into the Design History File, the Device Master Record, and the Device History Record is not known in ISO 13485.
- Conversely, the requirements of ISO 13485 and ISO 9001 for customer satisfaction and continuous improvement of the QM system go beyond the requirements of QSRs.
- Furthermore, the handling of complaints and the reporting system differ significantly.
Notes
The FDA does not recognize ISO 13485 certification as proof of conformity with the requirements of 21 CFR part 820. In contrast to ISO 13485, there is also no certification according to 21 CFR part 820.
Even before the harmonization of QSRs with ISO 13485, manufacturers can simultaneously demonstrate compliance with the requirements of 21 CFR part 820 and ISO 13485:2016 as part of the Medical Device Single Audit Program MDSAP.
3. Summary and conclusion
It is a great step forward for the harmonization of regulatory requirements that the FDA has essentially replaced its requirements for a QM system in 21 CFR part 820 with reference to ISO 13485. It explains in the "new" 21 CFR part 820 how certain requirements, e.g., for product identification and vigilance, are to be met in concrete terms.
It would have served medical device manufacturers if the MDR and IVDR had also followed this path.
Tip
Do you need support establishing or improving an ISO-13485 or/and FDA-compliant quality management system? Or do you need help with the changeover from QSR to QMSR? We are happy to help you!
Get in touch here.
Change history
- 2024-02-21: Revision based on the passed version of the QMSR from February 02, 2024

Author:
Luca Salvatore

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.