DAkkS: What's behind the outrage about ISO 13485?
The DAkkS, the German Accreditation Body, is Germany's national accreditation authority.
Recently, manufacturers, associations, and certification bodies have been raising their voices against the DAkkS. It is being vilified as an example of how German bureaucracy nips any innovation in the bud.
What is the truth of these accusations? Are they justified?
This article presents the work of the DAkkS and answers questions about the current situation in an FAQ.
1. The tasks of the DAkkS
The task of the DAkkS is primarily to ensure the safety, scientific reproducibility, and comparability of attestations of conformity. To this end, it monitors the conformity assessment bodies in the state accreditation procedure. Examples of confirmations of conformity are
- laboratory measurements,
- certificates from certification bodies, and
- calibration certificates.
The DAkkS assesses the technical competence, the scientific suitability of the test methods, and the impartiality of the bodies.
Through international agreements, DAkkS also ensures that accredited certificates or test reports are recognized outside Germany as well to simplify trade.
2. The responsibilities of the DAkkS
a) No responsibility for notified bodies for medical devices
In the area of medical device law, Germany is taking a special approach to quality assurance through notified bodies for the MDR and IVD. This is because Germany does not rely on accreditation by the DAkkS here but on designation by the ZLG.
The DAkkS is, therefore, not active in the area of conformity assessment of medical devices. In Germany, the ZLG is the sole competent authority for notified bodies and their subcontractors, including laboratories (see §§ 17-22 MPDG).
A notified body is organized in accordance with ISO/IEC 17065. It is both authorized and obliged to review all requirements of the MDR, in particular Article 10 of the MDR. This also includes the quality management system in accordance with ISO 13485.
b) Responsibility for certification bodies for management systems (ISO 13485)
Manufacturers or suppliers can also be certified voluntarily according to EN ISO 13485 and outside of the conformity assessment by a notified body for the approval of a medical device. Such a certificate could help to demonstrate the processes for compliance with the regulatory requirements for the organization (manufacturer, placing on the market, supplier), such as the requirements of Article 10 of the MDR.
However, this requires that
- these requirements are mentioned in the regulatory annexes ZA 1 to ZA 3.
- the notified bodies assume both the output from the certification of the devices in accordance with ISO/IEC 17021-1 and the responsibility.
Certification bodies that are not notified bodies but wish to issue certificates in accordance with EN ISO 13485 must themselves be accredited in accordance with ISO/IEC 17021-1. The DAkkS is responsible for this accreditation.
Note
This accreditation by the DAkkS must not be confused with the designation by the ZLG.
There are organizations that are "only" certification bodies and testing organizations that are both "certification bodies" and "notified bodies."
c) Further responsibilities
In addition to the certification bodies, the DAkkS is also responsible for the testing bodies/laboratories and the calibration bodies.
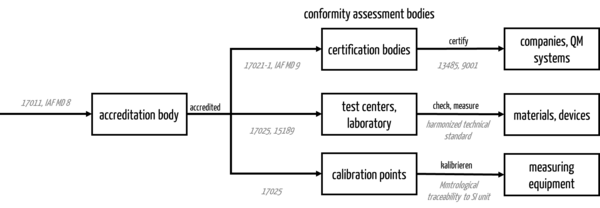
3. DAkkS: Regulatory categorization
a) Legal framework
The EU accreditation bodies are themselves subject to the regulatory requirements of Regulation (EC) 765/2008. EN ISO/IEC 17011 specifies these legal requirements.
All EU accreditation bodies regularly undergo a peer evaluation in accordance with Art. 10 of the above-mentioned regulation. The EA (European co-operation for Accreditation) organizes these evaluations. It ensures that the national accreditation bodies work in a harmonized manner, as far as required by Union law.
The DAkkS publishes the outputs of these evaluations here, for example, the results from 2022 (only available in German).
b) Mutual recognition
Within the EU/EEA, the mutual recognition of DAkkS accreditation and conformity confirmations of DAkkS-accredited laboratories and certification bodies follows from Art. 11 Para. 2 of Regulation (EC) 765/2008. This means that such certificates are valid throughout the EU internal market and the EEA.
If mutual recognition is to take place with third countries but within the WTO states, this must be agreed upon in EU reciprocity agreements under international law or in bilateral contracts with Germany. These agreements must explicitly refer to the MLA/MRA level of the International Accreditation Forum (IAF) or the International Laboratory Accreditation (ILAC). This is not the case in the area of medical devices and management systems for ISO 13485 (see FAQ 62 Medical Device Single Audit Program (MDSAP) and see question 9 below).
c) Special case management systems according to ISO 13485
Regulatory framework
The certification of management systems according to ISO 13458 is voluntary. There are no legally binding obligations for mutual recognition via the IAF-MLA. However, DAkkS has adopted the requirements of IAF for ISO 13485 for the area of voluntary certification in its administrative practice.
In particular, the documents IAF MD 8 for the concretization of ISO/IEC 17011 and IAF MD:9 for the concretization of the requirements of ISO/IEC 17021-1 apply to this area.
The accreditation bodies should at least request these. However, the documents have no external effect. They are neither standards nor laws. Neither manufacturers nor conformity assessment bodies can refer to them directly or are bound by them. Rather, they are administrative regulations that the accreditation bodies, as members of this organization, may adopt in their administrative practice.
Further information
The DAkkS maintains a list of adopted international rules (only available in German).
Effects
Whether the minimum requirements of IAF rules, EA rules, or ILAC rules are met only plays a role in the peer evaluation between the accreditation bodies. If minimum requirements are not met, this could lead to problems during the peer evaluation. Only in the specific situation must the evaluation team clarify whether the deviation meets the minimum requirements or not.
It is, therefore, permissible for stricter requirements to apply and be met in certain regions than those defined by the IAF or ILAC.
4. FAQ on the accreditation practice of the DAkkS
Question 1: What triggered the outrage about the DAkkS?
Since July 2023, the DAkkS has included an explicit description of the "regulatory scope" on the ISO/IEC 17021-1 certificate for bodies that certify to EN ISO 13485.
Many manufacturers and associations such as MedicalMountains fear (only available in German) that ...
"... already issued and valid certificates according to ISO 13485 would be immediately limited in their usability to the European Economic Union and Germany and [...] this would put manufacturers with a German certification body at a disadvantage inside and outside the EU".
There are also complaints about the DAkkS operating on its own.
Question 2: What has the DAkkS actually changed?
The certificates were amended ...
The DAkkS has redesigned the accreditation certificates since July 2023. Previously, they looked as shown in Fig. 2.

For the reasons stated below, the DAkkS has seen a need for clarification. It is now adding a detailed explanation of the scope of the "applicable regulatory" requirements, which are to be confirmed by the annex to "EN ISO 13485" under the addition "as described below" (see Fig. 3).

What is new is an explanation in chapter A1 of what the addition "EN" to "EN ISO 13485" means.
It is, therefore, actually the case that DAkkS has changed the presentation and describes the scope in more detail.
However, the specific accreditation confirmation still refers to the same harmonized standard, namely EN ISO 13485, in the applicable harmonized version. The confirmation was issued for the applicable Union law and not outside of Union law.
Note
According to the database of accredited bodies (in which the accreditation certificates can be retrieved), the DAkkS has always only issued accreditations for "DIN EN ISO 13485". No accreditation certificate can be found in which an ISO 13485 has been accredited without the addition "EN."
The DAkkS confirms that it has only issued accreditations for the EU/EEA legal area, which would be expressed by the indication "EN ISO 13485". This is because only the EN standard contains the regulatory requirements for ISO 13485:2016 in the EU.
... but the message remains the same
According to the DAkkS, both the old and the new certificates confirm exactly the same thing: a certification body accredited according to ISO/IEC 17021 can certify quality management systems according to EN ISO 13485. In other words, quality management systems that take into account the regulatory requirements for processes specified in the annexes of the EN version of ISO 13485. This is precisely the objective of the European harmonization of this standard.
In contrast to manufacturers and associations, the DAkkS, therefore, cannot recognize any change in practice. Rather, it speaks of a clarification.
Further information for interested readers
Accreditation, according to DIN EN ISO 13485, does not mean that the legal scope is limited to Germany. A manufacturer based in Switzerland or India who wants to deliver to the EU can also be certified for the requirements of EN ISO 13485.
The abbreviation DIN stands only for the German preface and the authorized German translation of the standard "EN ISO 13485", i.e., including the EU annexes.
The abbreviation EN means: The certification body can competently assess whether the manufacturer's QM system has implemented the regulatory requirements from Annexes ZA1 to ZA3 of ISO 13485, which contribute to the regulatory requirements of the MDR or IVD.
Question 3: Why did the DAkkS make this change?
The reason for this was a complaint from a national authority responsible for the approval of medical devices in the Middle East. It complained to DAkkS that a manufacturer had placed its devices on the market in this country without having the basic knowledge, necessary processes, and reporting channels of the medical device law applicable there.
This manufacturer had a certified QM system in accordance with "EN ISO 13485" under a German DAkkS accreditation and presented this certificate (without authorization) as proof of its competence.
In the opinion of the DAkkS, the subsequent disputes have shown that there is a need for clarification regarding the meaning of the term "EN" before the ISO 13485 standard is mentioned on the accreditation certificates.
Question 4: How does DAkkS justify the "regulatory scope" of accreditations?
ISO 13485:2016 differs from other management system standards according to ISO/IEC 17021 (such as ISO 9001) in its declaration of conformity. While other management systems only confirm conformity with the management system standard, ISO 13485:2016 explicitly requires that the certification also covers the conformity of the certified customer's processes for implementing the applicable legal requirements.
This can be seen from more than 20 references in the ISO 13485 standard, including the following examples:
”Several jurisdictions have regulatory requirements for the application of quality management systems by organizations with a variety of roles in the supply chain for medical devices. Consequently, this International Standard expects that the organization:
- identifies its role(s) under applicable regulatory requirements;
- identifies the regulatory requirements that apply to its activities under these roles;
- incorporates these applicable regulatory requirements within its quality management system.
The definitions in applicable regulatory requirements differ from nation to nation and region to region. The organization needs to understand how the definitions in this International Standard will be interpreted in light of regulatory definitions in the jurisdictions in which the medical devices are made available.“
ISO 13485, Kap. 01 „General“
Certification must ensure compliance with the following requirements:
“The organization shall document a quality management system and maintain its effectiveness in accordance with the requirements of this International Standard and applicable regulatory requirements.
The organization shall establish, implement and maintain any requirement, procedure, activity or arrangement required to be documented by this International Standard or applicable regulatory requirements.
The organization shall document the role(s) undertaken by the organization under the applicable regulatory requirements.”
Tz. 4.1.1 ISO 13485
“The organization shall (…) establish and maintain records needed to demonstrate conformance to this International Standard and compliance with applicable regulatory requirements (…).”
Tz. 4.1.2 ISO 13485
“The organization shall manage these quality management system processes in accordance with the requirements of this International Standard and applicable regulatory requirements.”
“Changes to be made to these processes shall be (…) c) controlled in accordance with the requirements of this International Standard and applicable regulatory requirements.”
Tz. 4.1.4 ISO 13485
“The organization shall retain responsibility of conformity to this International Standard and to customer and applicable regulatory requirements for outsourced processes.”
Tz. 4.1.5 ISO 13485
Consequently, if a certification body wants to certify an organization according to "EN ISO 13485", it must be able to assess whether the processes comply with ISO 13485 and any applicable regulatory requirements that are to be applied for placing on the market at the specific manufacturer and that are to be certified.
For this purpose, the certification body must employ personnel who have knowledge of the applicable regulatory requirements. Only in this way can it assess whether the quality management system of the customer to be certified correctly implements the regulatory requirements for the customer, the manufacturer/supplier (not the device!), and the necessary processes.
Question 5: Does this mean that DAkkS is restricting the scope to the EU?
Short version of the answer
No. The accredited certification bodies can serve customers based anywhere in the world and confirm to them through certification that they meet the requirements of EN ISO 13485 and thus the regulatory processes of the MDR and IVD in accordance with Annex ZA through their quality management system.
If the certification bodies wish (for their customers) to have a broader regulatory scope than the EU/EEA, they must demonstrate their competence (i.e. training of auditors) and apply for this scope.
Note
As shown above, notified bodies are not affected. They can certify in all legal areas in which they are notified, including the quality management system. Manufacturers who work with bodies notified by the ZLG or with bodies notified in accordance with MDSAP, for example, are therefore not affected.
Longer version of the answer
To date, the DAkkS has only issued accreditations to certification bodies for EN ISO 13485. However, it is also possible for German certification bodies, according to ISO/IEC 17021, to prove during the accreditation process that they employ personnel who are also familiar with regulatory systems outside the MDR/IVD, e.g., those of the "MDSAP countries" or Taiwan.
In other words, if a certification body according to ISO/IEC 17021 fulfills the requirements just mentioned, DAkkS can confirm its competence in other regulatory areas outside the MDR/IVD in the accreditation procedure.
Either there are separate standard annexes to ISO 13485 for some countries (e.g., for Canada: CAN/CSA ISO 13485:2016), or other regulatory requirements for the QM systems or processes or reciprocity agreements are applied for and confirmed directly (e.g., for the USA: 21 CFR Part 820 - Quality System Regulation; 21 CFR Part 803, 21 CFR Part 806, 21 CFR Part 807 - Subparts A-D; 21 CFR Part 821 - Device Tracking) (where applicable).
An unrestricted or unspecified "global" scope is not possible for the reasons mentioned above. This is because accreditation confirms the existence of a specific competence of the certification body.
Question 6: Is the DAkkS operating on its own?
With regard to the competence of personnel in certification bodies
The DAkkS denies this. It has adopted the worldwide minimum requirements of the International Accreditation Forum (IAF) for the ISO 13485 standard in its administrative practice (see above) so that manufacturers/suppliers certified under DAkkS accreditation can refer to the IAF MLA for ISO 13485. DAkkS, therefore, expressly confirms to the certification bodies on the accreditation certificates that they fulfill the requirements of ISO/IEC 17021-1 and also the additional international minimum requirements according to IAF MD 9.
In Mandatory Document (MD) 9, the IAF explicitly requires that the "Accreditation Bodies" (e.g., DAkkS) review that the CABs (Conformity Assessment Bodies), i.e., the certification bodies, have the necessary knowledge of the applicable legal framework (see line 2 Table B2 in Fig. 4).
In the case of CABs, this concerns both the auditors and the persons who check the audit results and decide on the issuing of certificates.
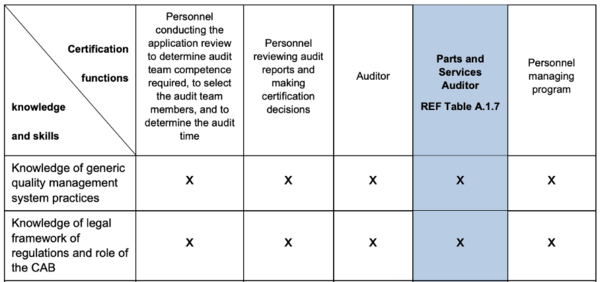
It can be concluded from this that all accreditation bodies worldwide are obliged under the IAF-MLA to review whether the accredited certification body also has the knowledge of the "applicable regulatory requirements" within the meaning of ISO 13485, which the certification body has explicitly requested.
Regarding the certificate
The DAkkS does not see a solo attempt here, either. Regulation (EC) 765/2008 obliges all EU accreditation bodies to comply with the requirements of ISO/IEC 17011 at least. This includes specifying the scope of application.
“The scope of accreditation shall, at least, identify the following. For certification bodies:
the standards, normative documents and/or regulatory requirements to which management systems, products, processes and services, or persons are certified, as applicable.”
7.8.3 ISO/IEC 17011
DAkkS complies with this European obligation by referring to the European harmonized standard "EN" ISO 13485, which is referred to as "the legal requirements" in its Annex ZA.
Note: There is no harmonized design of the accreditation certificates
In no economic sector are the accreditation certificates between the accreditation bodies exactly harmonized in their concrete presentation. Among other things, this is difficult due to the diversity of technology and the different requirements of the respective national administrative law, which is not harmonized at European or international level. However, the regulations in ISO/IEC 17011 and the IAF MD 8 for ISO 13485 define minimum information that all accreditation bodies must provide. In particular: All accreditation bodies place the "EN" or another abbreviation in front of ISO 13485 to describe the regulatory scope.
Question 7: What are the implications for medical device manufacturers?
The scope of a DAkkS accreditation certificate for ISO/IEC 17021 initially has nothing to do with the manufacturers. It is solely a question of which competence of a certification body according to ISO/IEC 17021 is confirmed by the DAkkS.
Impact on placing on the market in Europe
Placing medical devices on the market requires (depending on the class of these devices) the involvement of a notified body. Notified bodies are subject to the supervision of the ZLG and not the DAkkS.
There is, therefore, no effect on the market placement of medical devices in Europe. All manufacturers who work with a body notified by the ZLG are not affected. The designation by the ZLG does not change.
Effect on placing on the market in other countries
There is no direct impact on the placement of devices on the market in other countries because certification according to ISO 13485 by a certification body according to ISO/IEC 17021 is neither mandatory nor sufficient proof for the placement of medical devices on the market.
However, it may be useful for manufacturers to provide evidence of ISO 13485 certification in conjunction with applicable regulatory requirements if a notified body or medical device authority reduces its assessment program as a result. This could be of interest to countries participating in the MDSAP program, for example.
Again, a notified body participating in the MDSAP program is not affected by a change to the accreditation certificate according to ISO/IEC 17021. Countries participating in the MDSAP can take into account the output on the QM system according to ISO 13485 with the additional regulatory requirements.
Note
Regardless of the type of certification (either by a notified body or a certification body according to ISO/IEC 17021), manufacturers must and have always had to determine and comply with the legal requirements of the respective countries in which they wish to assume responsibility for placing on the market.
Otherwise, they must contractually transfer this risk and the legally required activities to a (local) manufacturer or buyer. In this case, the QM system only needs to describe and control the customer acceptance process so that the effectiveness of the contracts is ensured.
Impact on audits and certificates of certification bodies according to ISO/IEC 17021
The accreditation certificate concerns the legal relationship between DAkkS and the respective certification body.
Whether a certificate is withdrawn from a manufacturer is, therefore, not decided by DAkkS but solely by the certification body on the basis of the contracts between the manufacturer and the certification body.
For (legal) manufacturersin accordance with EN ISO 13485 by certification bodies in accordance with ISO/IEC 17021 (which are not notified bodies). This is because, according to the DAkkS, there has been no change in accreditation. The old certificates were issued for "EN" ISO 13485, just like the new certificates confirming "EN" ISO 13485.
There is no apparent reason to restrict or withdraw certificates, and according to DAkkS, no such reason has arisen to date.
More on notified bodies below.
Note
The certification bodies typically commit themselves contractually to the manufacturers to carry out accredited certification to EN ISO 13485. If the certification body discovers that a certificate is not yet covered by the scope of the accreditation certificate, it must submit an application for extension to the DAkkS. Otherwise, it runs the risk of not being able to fulfill its contractual obligations towards the manufacturer.
The certification body typically has no right to terminate the contract with the certified customer because it does not have the promised accreditation. It can happen at any time that the DAkkS identifies non-conformities at a certification body. The certification body is then obliged to take corrective action to restore the accreditation requirement and provide evidence of this to the manufacturer.
Manufacturers therefore do not have to do anything with regard to their certificates.
Question 8: What are the effects on suppliers?
The statement about manufacturers also applies analogously to suppliers. This means that suppliers do not have to do anything, either. However, their accredited certification body or notified body is contractually obliged to take all necessary and reasonable steps to provide the performance as contractually agreed under a valid accreditation or a valid designation.
Question 9: What are the implications for notified bodies?
No impact on the activity of notified bodies in the EU
(Legal) manufacturers who have their QM system evaluated in accordance with ISO 13485 by a notified body from Germany or the EU are not affected by the described change to the accreditation certificate.
Notified bodies that are organized in the EU according to ISO/IEC 17065 and have been designated by a "Medical Device Authority" such as the ZLG may perform several so-called "evaluation activities," e.g.:
- Inspection of devices or prototypes
- Evaluation of QM systems or production control
- Certification of the device on this basis, after which the manufacturer may place the device on the market
The problem of certification bodies according to ISO/IEC 17021 discussed here has no relation to confirmations by certification bodies according to ISO/IEC 17065 or by a "Medical Device Authority" notified body. This is due to the fact that it is either a different accreditation or no accreditation at all but a designation.
It follows from this: Even if a assessment company has both a designation by the ZLG and possibly another foreign "Medical Device Authority" and is also active in another business area as a certification body in accordance with ISO/IEC 17021, these are still completely separate legal areas. The activity as notified bodies is not affected by the accreditation according to ISO/IEC 17021 and vice versa.
No impact on the activity of notified bodies outside the EU
Manufacturers must comply with the regulatory requirements of the countries. In countries participating in the MDSAP program, manufacturers demonstrate conformity with ISO 13485 and the additional local regulatory requirements of the country in which they intend to place medical devices on the market. In order to avoid double assessments, this is accepted by other MDSAP states with regard to the statement of conformity to ISO 13485 and the MDSAP criteria, including the regulatory requirements. The EU does not participate in MDSAP.
Notified bodies that test medical devices, including the QM systems of these manufacturers, are - unlike certification bodies that only issue certificates in accordance with ISO/IEC 13485 for the QM systems - not accredited in accordance with ISO/IEC 17021 but in accordance with ISO/IEC 17065 or "designated" for e.g., MDSAP. According to the document "IMDRF MDSAP WG N3" and FAQ No. 62, there is no connection between an accreditation according to ISO/IEC 17021 under the IAF-MLA and the MDSAP program. The accreditation of notified bodies according to ISO/IEC 17021 by the DAkkS is therefore not relevant for the approval according to "MDSAP AS P0034".
Some assessment companies that are active in the EU as "Notified Bodies" have also been designated as "MDSAP Auditing Organizations." They may also issue "MDSAP certificates," which contain an assessment of whether the manufacturer also meets the QM requirements of these countries.
Manufacturers who have been certified that their device complies with ISO 13485 and the requirements of the participating MDSAP country have provided comprehensive proof for all MDSAP countries.
Question 10: What are the implications for certification bodies?
The DAkkS expects certification bodies, according to ISO/IEC 17021, to have the competencies required in IAF MD 9. It follows from this:
If the certification bodies request a broader scope than EU/EEA from DAkkS, they must provide evidence of the competence of their auditors through appropriate training certificates for the requested scopes. They must provide the authority with evidence that the regulatory requirements are known and understood by the auditors, as only then can they assess whether the QM system has implemented the processes effectively.
To this end, the certification bodies can, for example, use training courses for auditors participating in the MDSAP program for MDSAP countries. If the differences in the regulatory system are small compared to the EU, even internal training by the body may suffice (e.g., for Switzerland).
Applications for extensions can be submitted to the DAkkS accordingly.
Notes
If a certification body, according to ISO/IEC 17021, determines a need for action regarding the scope of application and subsequently submits an extension application, there is no need for further official measures. In particular, DAkkS will not withdraw any certificates (see above).
All German bodies may only be accredited by DAkkS. The "country of domicile principle" applies.
This could affect certification bodies that were previously only accredited in accordance with ISO/IEC 17021 for the scope "DIN EN ISO 13485" but have nevertheless issued certificates that did not merely confirm conformity with "DIN EN ISO 13485". DAkkS is not aware of any such case to date.
5. Summary and conclusion
a) The situation is assessed very differently
The assessment of the situation by manufacturers, certification bodies, and notified bodies on the one hand and DAkkS on the other varies:
- Manufacturers already feel sufficiently constrained by the MDR and IVDR, notified bodies, and the EU (with its MDCG documents and slow harmonization). Now there is the DAkkS. Many see this as an imposition. This is why manufacturers are instrumentalizing associations, ministries, and standardization organizations. A veritable shitstorm is brewing.
- The DAkkS cannot understand this because it has only made a clarification but has made no change.
b) Not everyone who complains is affected
Even if one interprets the clarification of the DAkkS on its accreditation certificates regarding the abbreviation "EN" as a change, not all organizations that are up in arms against it are affected:
- The accreditation certificates only affect the legal relationship between DAkkS and the certification bodies according to ISO/IEC 17021. Notified bodies and manufacturers are not affected.
- Manufacturers have always "only" had to determine, comply with, and provide evidence of compliance with the legal requirements for the countries in which they place their devices on the market. This means that no change can result from the certification of the QM system.
c) What can we learn from this?
Perhaps at least something can be learned from this (unnecessarily?) stressful situation:
1. Talk to each other
Professional communication with each other and not about each other helps to prevent or at least contain negative emotions, escalations, and legal disputes. This requires prompt and binding responses to inquiries.
2. Use the risk-based approach
The benefit of regulatory requirements for patient safety and care does not scale with the number of regulatory requirements. The effort required to meet these requirements does. Therefore, the risk-based approach should also apply here.
If a manufacturer does not know how it can and must recall unsafe medical devices quickly and effectively in a country, this represents a high risk. It, therefore, makes sense for manufacturers to (also) know and comply with the vigilance requirements of the respective countries. And that the certification bodies or notified bodies (can) also check this.
3. Do not forget the patients
We have long observed that manufacturers are no longer offering affordable medical devices to the same extent in order to diagnose and treat as many patients as possible. This is a problem for all of us because we are all potential patients. For example, one very large manufacturer has halved its portfolio. Not only older devices were removed, but also devices for children and for patients with rare diseases.
In other words, even if everyone is right in the end, no one is helped.
4. Simplify the system
The regulatory system has become so complicated that this further extensive article was needed to shed light on just one aspect.
We have tried to shed more light on the subject with the information and FAQs presented above. We have asked the DAkkS for comments on individual questions and received answers, which we have taken into account accordingly.
Overall, the problem appears to be less problematic from the manufacturer's point of view than initially assumed. Notified bodies are not affected, and certification bodies, according to ISO/IEC 17021, are only affected in certain foreign constellations outside the EU.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.