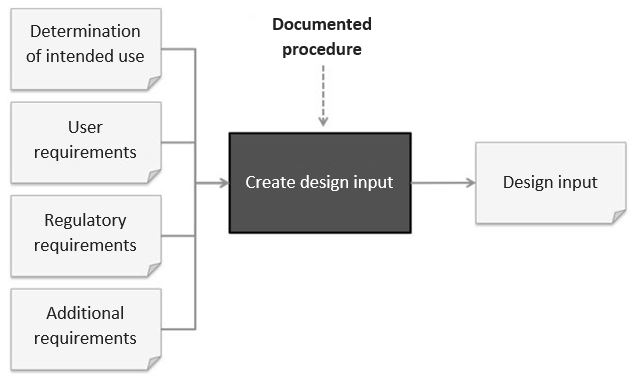
Design input: What you shouldn’t forget
“Design Input" refers to the development specifications, and it's not just the FDA that makes concrete demands in this regard.
This article describes what content your design input should contain. You will learn how risk management interacts with the design input.
Definition of the term “design input”
The FDA defines the term as follows in 21 CFR 820.3(f):
Definition: Design input
"Design input means the physical and performance requirements of a device that are used as a basis for device design."
Source
ISO 13485 also uses this term, but without defining it.
Regulatory requirements
a) FDA 21 CFR 820.30
The FDA also defines the requirements for the design input. It states:
Each manufacturer shall establish and maintain procedures to ensure that the design requirements relating to a device are appropriate and address the intended use of the device, including the needs of the user and patient. The procedures shall include a mechanism for addressing incomplete, ambiguous, or conflicting requirements. The design input requirements shall be documented and shall be reviewed and approved by a designated individual(s). The approval, including the date and signature of the individual(s) approving the requirements, shall be documented.
b) ISO 13485 chapter 7.3.3 (“Design and development inputs”)
ISO 13485 expects the following to be part of the design and development inputs:
- Function, performance and safety requirements so that the product can be used correctly for its intended use
- The regulatory requirements
- If applicable, requirements derived from previous developments
- Requirements resulting from risk management and
- Other requirements (without specifying them in more detail).
Like the FDA, ISO 13485 also requires the product requirements to be determined, evaluated and documented.
c) Changes in ISO 13485:2016
ISO 13485:2016 has increased the design input requirements.
Usability requirements
The design inputs must now also include the “usability requirements”. The standard does not define what “usability requirements” are. They could include:
- User requirements i.e. what a user must be able to enter, select or recognize using the system
- Design specifications like the ones you can find, for example, in style guides. Examples are contrast ratios, font sizes and positions.
Verifiable or validatable design inputs
The previous version of the standard already required the design inputs to be unambiguous, consistent, complete and appropriate/correct, and for this be verified in a review.
ISO 13485:2016 requires that the requirements must also be validatable or verifiable (i.e. testable on the product). This should be self-evident, but it is now explicitly required.
Design and development file
The design inputs must be part of the design and development file. This file was introduced in ISO 13485:2016 and corresponds to the FDA's “design history file”.
Further Informationen
Read more on the subject of the design history file here.
Design input content
Because companies often do not clearly differentiate between the Intended use, needs, Stakeholder requirements and System requirements, they are all grouped under design input. After all, the “design” should take everything into account. But that’s not right.
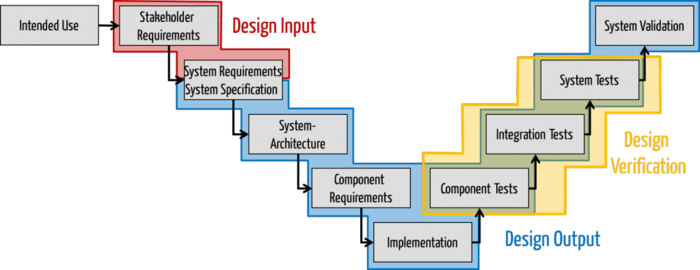
Intended use and needs: not part of the design input
The intended use is not part of the design input. The FDA explicitly requires a process to be documented detailing how the “design requirements” (which it uses as a synonym of “design input”) are drawn up so that this intended use can be achieved.
The user needs are not yet part of the design input according to the FDA. Instead, the design input must be suitable for meeting these needs.
Stakeholder requirements: part of the design input
ISO 13485 explicitly includes the regulatory requirements, which are part of the stakeholder requirements, as part of the design inputs.
System requirements and system specification
The term system requirements specification contains two separate terms:
- The system requirements: Requirements for a system (often “under the hood”)
- The system specification: Definition of how a system should be designed, e.g. the UI specification (“above the hood”, i.e. the visible part)
The system requirements should be included in the design input. Finally, the product requirements are determined and these are, by definition, part of the design input.
The system specification (above the hood) can be included in both the design input and the design output:
- On the one hand, you can argue that the UI specification determines what the product looks like. This specification could be interpreted as product requirements and therefore as a design input.
- On the other hand, you can argue that the UI specification is already a result of development and is, therefore, part of the design output.
We recommend the second option because the UI specification is indeed a result of development.
Caution!
Even if the UI specification is part of the development, it should not be the job of the developers to create it, but instead it should be the job of usability engineers, especially interaction designers.
If you don't distinguish between system requirements and system specifications, then include the system requirements and software requirements specification (SRS) in the design input.
Thanks to Dr. Markus Bentrup for his help in clarifying this.
Changes to the design input
During development, you will need to revise the design input and the design output accordingly. Make sure that all versions of these documents become part of your design history files. This is the only way to document the development history in a traceable and credible way.
Risk analysis as design input
Risk management badly done
We often see medical device manufacturers using the following procedure:
- Build the medical device as safely as possible
- Perform the risk analysis
- In the event that unacceptable risks are discovered, either sweep these under the carpet or make the medical device safer (e.g. a different system architecture)
- Verify these measures and document everything in the risk management file.
If you use this procedure you run the risk of wasting a lot of time and money.
Better: Risk analysis as design input
The result of the risk analysis should itself become part of the design input.
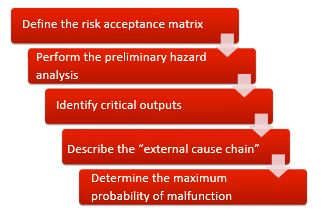
We suggest the following procedure:
- Define the unacceptable risks quantitatively. (You can learn how to do this in three short videos on Auditgarant).
- Carry out a preliminary hazard analysis and identify the hazards.
- Find out from which hazards are caused by which of your device’s outputs (can include the UI).
- Describe how the output can lead to the hazard (if the output is not the hazard itself) and consider the likelihood of the hazard occurring in the event of a hypothetical medical device output malfunction.
- Now determine, using the risk acceptance matrix, the maximum probability of an output functioning incorrectly. This maximum probability is now the design input.
Only now develop your medical device according to this design input. For example, if the design input specifies a probability that a group of (electronic) components or self-written software cannot guarantee, you will have to choose a different system architecture, e.g. a diverse system architecture.
Unfortunately, we almost never see design inputs (i.e. system requirements) that are a response to specific probabilities. The consequences are costly improvements, delayed projects or/and unsafe medical devices. You can avoid these problems!

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.