Qualification and classification of IVD software
The qualification and classification of IVD software determine how and how quickly IVD manufacturers can bring their software to market and what costs arise for "approval."
This article will help you correctly qualify and classify IVD software, thereby avoiding regulatory problems and the resulting costs and delays.
1. Definition of terms
1.1 Qualification
Qualification is the assessment of whether or not a device falls under the definition of a medical device or in vitro diagnostic medical device (IVD).
Note
The second chapter of this article describes the qualification of IVD software.
1.2 Classification
The term classification is used twice:
1. The risk classification of IVD refers to the categorization into classes A, B, C, or D according to Annex VIII of the EU Regulation 2017/746 on in vitro diagnostic medical devices (IVDR).
2. There is also a software safety classification according to the software standard IEC 62304, which distinguishes between classes A, B and C.
Note
The third chapter of this article describes the classification of IVD software, the fourth the safety classification according to IEC 62304.
1.3 IVD software
There is no universal definition of the term IVD software. In the context of this article, we understand IVD software to be
- a standalone software application,
- that qualifies as a standalone IVD (see chapter 2),
- which is an accessory to an IVD, or
- a software system that is part of an IVD instrument.
IVD software does not include standalone software applications that are used in the IVD context but which
- fall within the scope of the EU Medical Device Regulation MDR or
- are neither a medical device nor an IVD.
1.4 Medical Device Software – MDSW
The MDCG Guideline 2019-11 defines the term medical device software:
Definition: Medical Device Software
“Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the medical devices regulation or in vitro diagnostic medical devices regulation.“
Source: MDCG 2019-11
For software to qualify as medical device software (MDSW), it must have a medical purpose.
2. Qualification of software as IVD
The MDCG 2019-11 Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 –MDR and Regulation (EU) 2017/746 – IVDR is a valuable guide to the qualification of medical and non-medical software. The guidance document breaks down the qualification into two steps:
1. determination of whether the software counts as MDSW
2. determination of which EU regulation (MDR or IVDR) the software or device falls under
The following subchapters present these two steps.
2.1 Determination of whether the software counts as MDSW
In the first step, manufacturers must check whether the software falls under the definition of "medical device software." They can rule this out if one of the following conditions applies:
- The software does nothing other than store, archive, communicate, or offer a simple search for data.
- The software is not intended to provide a (medical) benefit to individual patients.
However, if the software (whether standalone or part of an instrument) is used for the diagnosis, therapy, monitoring, or prediction of diseases and injuries, it is, by definition, medical device software.
This means that the software or the device of which the software is a part counts as a medical device or IVD medical device and falls under the scope of the MDR or IVDR.
Further information
The MDCG 2019-11 guidance describes this first step in detail with the help of an illustration and explanations.
2.2 Determination of which EU regulation the software falls under
Whether the software or the entire device must meet the requirements of the MDR or the IVDR depends on several criteria:
- intended purpose of the device
- data sources for the information displayed or processed
- dependence of the intended purpose on these data sources
The second decision tree in the MDCG 2019-11 guideline explains the distinction between cases (see Fig. 1).
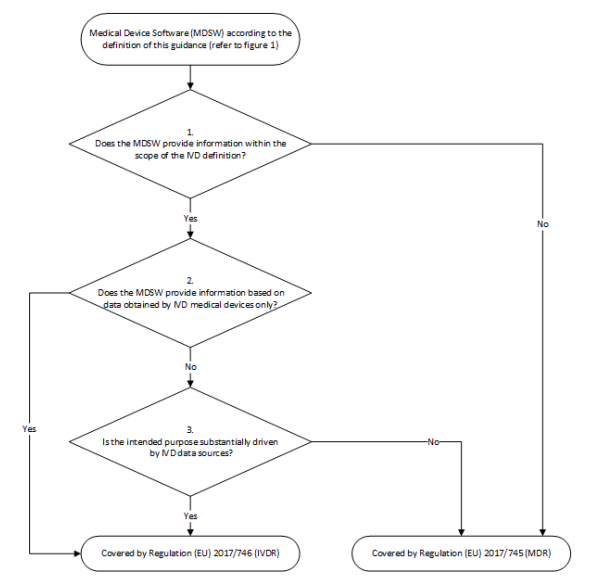
- The first question examines whether the software qualified as MDSW falls within the scope of the IVDR based on the IVD definition. If the answer is no, the software must be considered exclusively under the MDR. If the answer is yes, continue with question 2.
- If the software falls under the IVD definition, the second question is whether the input data originates exclusively from an IVD. If the answer is yes, the software falls under the IVDR. If the answer is no, we move on to question 3.
- The third question examines whether the software uses data from "IVD sources" as well as from other sources (e.g., medical devices). The aim is to find out whether the software's intended purpose is essentially achieved by IVD data sources. If the answer to this question is yes, the software falls under the IVDR. If the answer is no, the software falls under the MDR.
2.3 Further interpretations of the MDCG document
2.3.1 Labeling of laboratory values, e.g. "out-of-range" values
A system such as a laboratory information system (LIS) does not become an MDSW by the fact that it
- labels laboratory values, for example, to mark "out-of-range" values,
- filters laboratory values or
- forwards data to other systems.
Complex rules and calculations usually serve medical purposes and lead to qualification as a medical device or IVD medical device.
2.3.2 Parameterization and scripting
If manufacturers allow users to parameterize and possibly even scripting that has a medical purpose, the software (or at least the respective module) becomes MDSW, if this was not already the case. The organization that makes these adjustments thus becomes an in-house IVD manufacturer.
However, simple calculations such as unit conversions do not yet require software to be qualified as a medical device or IVD medical device.
Further information
Read here what health institutions such as medical laboratories must consider when manufacturing and developing in-house IVD, also called laboratory-developed tests (LDT).
Please refer to the EU Borderline Manual for further information.
3. Risk classification of IVD software according to IVDR
The IVDR distinguishes between classes A and D for devices qualified as IVDs. Class A comprises the non-critical devices, and class D the highly critical devices.
3.1 The effects of risk classification
The classification does not affect the safety and performance requirements the IVD medical device software must meet. Even for Class A software (not to be confused with Class A according to IEC 62304), all applicable requirements according to Annex I of the IVDR must be met. These include, among others:
- software life-cycle (IEC 62304)
- risk management (ISO 14971)
- usability (IEC 62366-1)
- IT security
- verification and validation
However, the classification determines the possible conformity assessment procedures: For Class A IVD medical devices, manufacturers do not have to involve a notified body, i.e., there is no inspection of the documents by a notified body. However, the competent authority can request access to the technical documentation during market surveillance.
Although manufacturers also need a QM system for Class A IVD medical devices, this does not have to be certified. The assessment of the QM system for class B to D IVDs is carried out automatically during the conformity assessment in accordance with Annex IX of the IVDR.
3.2 Classification rules according to IVDR
Similar to the MDR, the IVDR contains the following classification rules, among others:
1.1. Application of the classification rules shall be governed by the intended purpose of the devices.
1.2. If the device in question is intended to be used in combination with another device, the classification rules shall apply separately to each of the devices.
1.3. Accessories for an in vitro diagnostic medical device shall be classified in their own right separately from the device with which they are used.
1.4. Software, which drives a device or influences the use of a device, shall fall within the same class as the device. If the software is independent of any other device, it shall be classified in its own right.
1.8. Where a manufacturer states multiple intended purposes for a device, and as a result the device falls into more than one class, it shall be classified in the higher class.
1.9. If several classification rules apply to the same device, the rule resulting in the higher classification shall apply.
Source: IVDR, Annex VIII
The IVDR classifies devices, taking into account the underlying clinical conditions or the potential (critical) impact that incorrect information provided by an IVD can have on a patient/person. Therefore, there is no need for a "Rule 11" as in the MDR.
3.3 Application of the classification rules
Since the classification is based on the intended purpose, the decisive factor is whether the standalone software provides medical information on the patient, i.e., whether it allows a clinical statement to be made. In other words, whether the software first establishes the specificity and, if applicable, the sensitivity of the analyte.
Example
Bioinformatic software that compares genetic data from high-throughput sequencing of DNA samples with reference data, for example, to
- determine whether a disease is present or
- diagnose or analyze a specific physiological condition or
- classify stages of a cancer disease
fall into class C unless it is a "life-threatening disease with a high or suspected high risk of spreading" caused by transmissible pathogens. Then, the IVD software even falls into class D.
3.3.1 IVD software in "risk class" A?
The statement that IVD software always falls into Class A because software can never directly harm patients or because the software itself cannot do anything is not correct.
If software controls an IVD instrument, it is assigned to the same class as the instrument (according to implementation rule 1.4).
If the instrument complies with rule 5b) and does not perform any analyte-specific data evaluation, it falls into class A. Only in this case does IVD medical device software also fall into class A.
3.3.2 Risk classes B, C and D
If the IVD software performs assay-specific evaluations, it must be assigned to the class of the assay. The software influences the application and provides diagnostic information for the intended medical purpose.
If different assays are to be evaluated with the software, the implementation rules 1.8 and 1.9 must also be applied (classification in the highest class of the assays concerned!).
Caution
It is a strategic decision whether to separate software into driving modules and analyzing modules so that they can be classified differently.
Benefit from the Johner Institute's team of experts when defining your software segregation and regulatory strategy!
4. Safety classification of IVD software according to IEC 62304
4.1 Problem definition
A point-of-care device measures "common" parameters such as pH, pCO2, glucose, potassium, etc. How should its software be classified according to IEC 62304?
- One team argues in favor of safety class B because no serious injury is possible. After all, the measurement result of the IVD medical device is only one of many parameters for a therapy - the right one, the wrong one, or a correct but delayed one. In addition, calibration and control cycles are run regularly in the laboratory instrument.
- Another team argues in favor of safety class C because, in the unlikely event, there could actually be patient harm. They do not consider doctors a "risk-minimizing measure."
Which assessment is correct?
4.2 Specific characteristics of IVD
For IVD medical devices, or more precisely their output, unlike other medical devices, there is no possibility of direct harm: no crushing, no radiation damage, no electric shock. The measured values can "only" lead to patients being treated incorrectly, delayed, or not treated at all - which can have fatal consequences depending on the context of the application.
This "external chain of causes," i.e., the chain of causes starting with the incorrect, delayed, or non-displayed measured value, is difficult to estimate. The probabilities are particularly difficult to estimate - for example if a doctor actually treats incorrectly due to an incorrect or missing measured value.
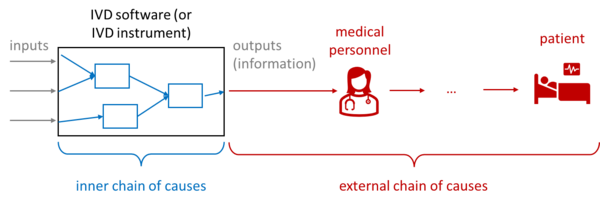
Unfortunately, IEC 62304 does not take these probabilities into account. This means that in the worst-case scenario, serious harm can occur in "sufficiently critical" patients, resulting in safety class C. IEC 62304:2006 + A1:2015 provides more options here through measures outside the software.
The calibration and check cycles usually do not change the severity; they only reduce the probability that a measured value is displayed incorrectly or not at all, leading to harm. However, probabilities are largely irrelevant for safety classification.
4.3 The intended purpose decides
The safety class for IVD medical device software, like the risk class (see Chapter 3), depends primarily on the intended purpose. This is because the intended purpose, in turn, determines the target patient population, i.e., the patients or their state of health or disease. This results in the maximum severity of harm that can be caused by a software failure and - largely independent of the probability - the safety class of the software in accordance with IEC 62304.
The intended function of the IVD medical device software also influences the target population and must be considered (e.g., a screening test in a predominantly healthy population or a confirmatory test based on risk factors or preliminary examinations and thus on a predominantly already diseased (high-risk) population). Finally, the performance parameters (such as the positive and negative predictive value) in the corresponding population show the probability that a person being tested will also receive the correct output.
In addition, the state of the art in medicine should be taken into account to assess the external causal chain leading to harm to the patient, to determine which treatment options are available and which patient management decisions are made based on the information provided by the IVD software.
The answer to the question in chapter 4.1. is, therefore, the intended purpose decides.
Further information
Read more about Software as Medical Device (SaMD) here.
5. Summary and conclusion
The qualification and classification of IVD software determine the duration and effort required for the "approval" of these devices.
However, manufacturers can influence the qualification and classification according to the intended purpose and the division of the device into one or more IVD medical devices, accessories, or non-IVD products.
These determinations are part of the regulatory strategy, which also influences approval in other countries and the post-market phase. Accordingly, the regulatory strategy is important and groundbreaking.
Support with regulatory strategy
The Johner Institute supports IVD manufacturers in wisely defining their regulatory strategy, avoiding unnecessary regulatory burdens, and enabling devices to be placed on the market as soon as possible.
Change history
- 2024-03-26: Article completely rewritten
- 2019-10-30: First version of the article published (due to MDCG 2019-11)

Author:
Dr. Tina Priewasser

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.