Post-Market Surveillance, Market Surveillance & Vigilance
Post-market surveillance (PMS) is defined as "a systematic process to derive necessary corrective and preventive actions (CAPA) from information on medical devices already placed on the market".
Content overview
- Objectives of the Post-Market Surveillance
- Distinction Between PMCF and Vigilance
- Regulatory Requirements
- Tips for Procedural Instructions
ISO 13485:2016 and ISO 14971:2012 require post-market surveillance. Moreover, the FDA published a revised guidance document in May 2016.
Update: MDR's requirements have been amended. Distinction between post-market surveillance and post-market clinical follow-up further specified.
Definition of Post-Market Surveillance
Both the Medical Device Regulation (MDR) and the FDA define the term "post-market surveillance":
Definition: Post-Market Surveillance
„all activities carried out by the manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from their devices placed on the market, made available or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions“
Source: MDR
The FDA's definition is comparable:
Definition: Post-Market Surveillance
„The active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device.“
Source: FDA
The FDA's definition may be conveniently short. However, the MDR's more long-winded explanation seems to be more helpful as it not only describes actions but also objectives of post-market activities.
Post-Market Surveillance: Objectives
Before bringing medical devices to market, manufacturers must minimize their risks and ensure patients' safety. This is monitored by authorities and notified bodies during authorization and conformity assessment procedure.
However, some risks manifest over time, when medical devices are used daily by practitioners.
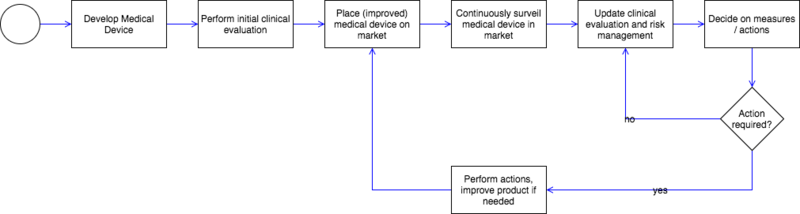
Post-market surveillance aims at
- systematically identifying these risks when occurring during practical usage
- monitoring the products' performance afield
- detecting product faults and safety issues left undetected
- constantly updating the benefit-risk assessment
- quickly initiating necessary measures such as recalls
Only due to a constant systematic market surveillance (post-market surveillance) can manufacturers guarantee that medical devices provide the promised benefit to patients as well as the lack of any unrestrained risks.
Please read below which data you can considered for market surveillance.
Distinction to Post-Market Clinical Follow-Up and Vigilance
Closely connected to post-market surveillance are the terms "post-market clinical follow-up" (PMCF) and vigilance.
Post-Market Clinical Follow-up (PMCF)
As already explained, post-market surveillance aims to continuously verify the benefits of medical devices and to identify previously unknown risks by observing and analyzing daily practical usage. If "regular market surveillance" does not provide sufficient data, post-market clinical follow-up studies regarding the manufacturer may become necessary.
If the PMS reveals information requiring changes to be made to the clinical evaluation, an update must be made. PMCF studies shall collect clinical data with the immediate aim to update and enhance clinical evaluations.
Definition: Post-market clinical follow-up (PMCF)
„A continuous process to update the clinical evaluation referred to in Article 49 and Part A of this Annex [XIII]“
Source: MDR
Hence, post-market clinical follow-up (PMCF) is about systematically collecting clinical data to answer important questions regarding the safety or performance of medical devices which have been left unsolved.
Post-market surveillance involves collecting all kinds of meaningful practical information, for example also in form of service reports, hotline calls, customer complaints etc.
PMCF aims at updating clinical evaluation. The objective of post-market surveillance is to decide on necessary measures guaranteeing the safety of patients and practitioners. The decision takes the results of clinical evaluation into consideration. Therefore, PMCF is a subset of post-market surveillance.
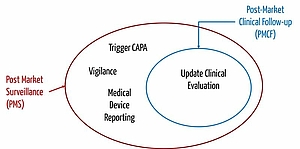
The MDR regards post-market clinical follow-up as part of post-market surveillance. The MDR says, for example, that
The manufacturer shall undertake to institute and keep up to date a post-market surveillance plan, including a post-market clinical follow-up
However, the FDA does not differentiate between the two aspects as precisely as the MDR: The FDA requires the post-market surveillance plan to include data determined when conducting studies, such as the participant number, the study objective and declarations of consent.
Vigilance
A vigilance system refers to a market surveillance and reporting system. During vigilance, manufacturers must not only regulate how they surveil the market, but also how the report incidents to the responsible authority. Laws and regulations such as the MPSV leave manufacturers little scope for specifying the reporting system.
Conclusion
The activities within the scope of post-market surveillance, market surveillance, reporting, post-market clinical follow-up and vigilance are partly overlapping. Thus, the terms are often used as synonyms - although they aren't.
Regulatory requirements usually refer to several aspects.
Regulatory Requirements
a) Medical Device Directive (MDD, 93/42/EWG)
The Medical Device Directive (MDD) explicitly requires (Appendix IX, clinical evaluation): manufacturers must update the clinical evaluation with data coming from post-market surveillance. They must justify if this process does not involve post-market follow-up.
b) Medical Device Regulation (MDR)
The new Medical Device Regulation, which will replace the MDD from mid-2017 on, offers a way more detailed and specific description of the requirements put on post-market surveillance. As many as four articles and an appendix are dedicated to this topic.
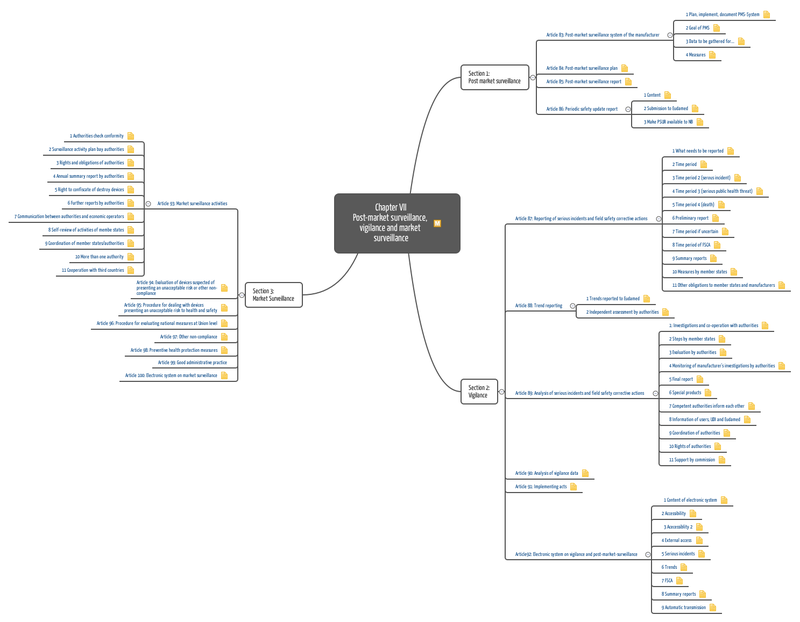
Important requirements of the MDR regarding post-market surveillance are:
- defining, planning and maintaining the post-market surveillance process
- continuously and systematically collecting and evaluating data
- based on these data, continuously decide on measures such as
- initiating CAPA (possibly referring to the product as well as the manufacturer and his processes)
- informing authorities and users
- initiating recalls
- updating clinical evaluations
- reporting on results (periodic safety update report)
Appendix III lays down which sources must be analyzed and how they must be assessed.
c) ISO 13485:2016
ISO 13485:2016 obliges manufacturers to guarantee the effectiveness of QM systems a medical devices' safety amongst other through systematic market surveillance (post-market surveillance).
d) ISO 14971:2012
The norm on risk management too lays down requirements about the "subsequent phase". However, the norm's focus is not reporting. Rather, the norm is about using information coming from production or following stages (e.g. products' usage) to assess
- if probability and severity of possible damages are accurately judged
- if risks are fully identified
- if presumed risk acceptance criteria and risk-benefit ratios are valid
Manufacturers acting under ISO 14971 must not only search for problems in the subsequent phase. All information aiding to verify or falsify the own assumptions' correctness is significant.
Please read below which data you can consider for market surveillance.
The risk management report must confirm that the planned activities are appropriate.
e) USA/FDA: 21 CFR part 822 and “Post-Market Surveillance” Guidance Document
In 21 CFR part 822, the FDA provides precise regulations on:
- When post-market surveillance is necessary
- What manufacturers need to consider in their post-market surveillance plan
- Which documents the manufacturers have to produce
- How quickly manufacturers must react in the event of a problem or the FDA must act
On May 16, 2016, the FDA published a Guidance Document specifically on 21 CFR part 822 that was intended to give manufacturers additional guidance. A new draft of this Guidance Document was published in 2021.
Additional information
Further information on the FDA requirements can be found in this article on 21 CFR part 822 and in the aforementioned Guidance Document.
f) IMDRF / GHTF
The International Medical Device Regulators Forum (IMDRF) has adopted documents from the defunct Global Harmonization Task Force (GHTF) which incorporates guidelines mainly regarding reporting.
g) MPG, MPSV
Although the MPG and MPSV demand a market surveillance, they impose specific requirements only on reporting and recalls.
h) USA /FDA: 21 CFR part 822 and Guidance Document "Post-Market Surveillance “
The FDA lays down provision precisely governing
- when a post-market surveillance is required,
- what manufacturers must keep in mind when planning market surveillance (post-market surveillance)
- which documents manufacturers must produce and
- how quickly manufacturers and the FDA must react in the event of an incident.
On May 2016, the FDA has issued a Guidance Document particularly on 21 CFR part 822 which offers manufacturers further assistance.
i) Further information
Please find further information on the FDA's requirements in this article on 21 CFR part 822 and the mentioned Guidance Document.
Conclusion
The European legislation on reporting (especially MEDDEV 2.12-1 and MPSV) is so extensive, specific and detailed. Therefore, manufacturers have little leeway when formulating respective process instructions.
However, European provisions on the actual market surveillance have not had as much impact on decision making so far - in contrast to the FDA's requirements. Now this will change due to the MDR with its much more comprehensive, precise and in-depth description of post-market surveillance. The following detailed tips assist you with implementation.
SOP "Post-Market Surveillance"
As a manufacturer of medical devices, you should structure your post-market surveillance, e.g. through respective standard operation procedure SOP, subject to the following aspects:
- Activity Triggers
In general, there are two possible triggers- based on a point in time or period (e.g. once every quarter, every third Monday of a month)
- occasion-related triggers (e.g. customer calls, a new norm was published, 10,000th device was shipped)
- Sources of Information
Examples for sources of information are- customer feedback including customer complaints
- service reports
- assessment results
- observations of employees
- data bases of authorities with reports on problems or measures by manufacturers of comparable products, technologies or techniques
- calls to the hotline
- specialized clinical literature
- results of post-market clinical follow-up studies
- fairs and conferences
- Activities
Activities typically involve- collecting data
- analyzing data
- evaluating data
- taking actions (e.g. recalls, reporting to authorities, CAPA) or justifying inactivity.
- Responsibilities
Process instructions should also describe which activities are assigned to which roles. For example, medical product advisors, safety officers, risk managers, developers, support specialists, hotline services and management are involved. - Documentation and Tools
As manufacturers evaluate comprehensive data when conducting post-market surveillance, we recommend documenting this information using tools. Do not forget to validate these tools, which is required under ISO 13485:2016.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.