Design History File: What Your DHF should Include
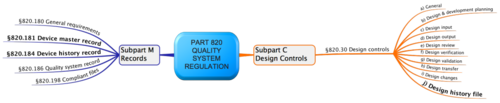
The FDA requires in 21 CFR Part 820.30 a Design History File DHF (these are the "Quality System Regulations"). DHF should not be confused with the Device History Record DHR or the Device Master Record DMR. This article explains what the Design History File must contain and how it differs from the other two artifacts.
Design History File
The FDA requires in 21 CFR part 820.30j (Design History File) that "Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records Necessary to demonstrate the design did what developed in accordance with the approved design plan and the requirements of this part."
Documents that should be included in a Design History File are:
- Design inputs such as a system or software requirements specification
- Design outputs such as system or software architectures or component drawings, as well as the risk analysis and risk assessment of the design.
- Design verification and design validation for example results of component, integration and system tests and results of usability verification and validation activities that should take place both during and at the end of development.
- Design transfer, which shows how the product has to be produced later.
For software, the FDA document "Content of the Premarket Submissions for Software Contained in Medical Devices" also gives you more information about the contents of the Design History Files.
The design history file cannot just represent the last stage of development. Rather, the Design History File must document the course of development comprehensibly, for example how changes Design Changes (changes to the design) have been incorporated, released and implemented. The Design History File thus corresponds to the history of the development documentation.
Your Design History File must prove that you, the manufacturer, have been compliant with your own standard operating procedures and work instructions, especially in respect to development.
Device Master Record DMR
Once the Design History File shows the device to be developed, the Device Master Record describes how the device is to be produced accurately. These requirements may include:
- how the device is to be assembled
- how production machines must be set
- how the device must be checked
- how the device is to be packed
- how the device must be installed and maintained
- etc.
The Device Master Record can be relatively short for standalone software.
Device History Record DHR
The Device History Record DHR finally provides evidence that one has produced the device in accordance with the requirements of Device Master Record and that it meets the requested acceptance criteria.
This "file" includes records such as the results of product testing, for example, at the end of production. Each unit (with the pharmaceutical products this would be each "batch"), is to be identified. This file must also include maintenance reports and results of installation tests.
For standalone software the Device History Record will be slim, since there is no real production. However, the reproduction of data media or the distribution over downloads count towards the file.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.