Medical Device Regulation MDR
As of May 25th 2017, the Medical Device Regulation MDR and In-vitro Medical Device Regulation IVDR entered into force. These regulations replace the EU directives (MDD, IVDD, and AIMD). As of May 26th, 2020, the MDR shall apply, on May 26th, 2022, the IVDR.
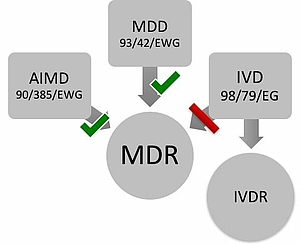
About this article MDR
This article gives on overview on:
- Changes by MDR (in comparison to MDD)
- Main differences to US / FDA regulations
- The Medical Device Regulation itself (MDR at a glance)
- Estimated time line and transition period
- Assessment and criticism
Medical Device Directive: What remains unchanged?
The changes are significant. Nevertheless, some core concept remained unchanged:
- No regulatory authority/agency: There won’t be a European agency granting market access comparable to the European Medicine Agency EMA for drugs or the FDA for drugs, medical devices and more.
- Manufacturers still declare conformance (with the CE mark) after following a conformity assessment procedure. The conformity assessment procedures still depend on the class of the devices. With exception of class I devices, notified bodies still have to be involved.
- The classification essentially is unchanged. Exceptions are mentioned below.
- Harmonized standards remain a way of proofing compliance. However, the EU additionally introduces the concept of “common specification” (see below).
- Non-EU-manufacturers can import medical devices. One of the prerequisites still is an EU representative.
MDR: What is new?
The Medical Device Regulation is a document spanning more than 550 pages – a multiple of the Medical Device Directive MDD. The MDR introduces numerous new requirements and concretizes requirements of the MDD.
- Essential requirements
The essential requirements – now renamed as “essential safety and performance requirements” – are much more specific. Consequently, the technical documentation is closer regulated. There is a comparision between MDD and MDR requirements. - UDI & Labeling
Like the FDA, manufacturers now must assign a unique device identification (UDI). This UDI consists of a device identifier and a production identifier. The UDI is not the only change to the labeling requirements. - New roles
Manufacturers must employ (exception for small companies) a “person responsible for regulatory compliance”. Other "economic operators" include roles such as the distributor, the EU representative and the importer. - EUDAMED
Not only the UDI data must be submitted to the EU database (EUDAMED), but also (dependent on the class) post-market data (e.g. a “periodic safety update report”) . - Clinical Evaluation and PMCF (post-market clinical follow-up)
The MDD was addressing the clinical evaluation only on one page and the post-market clinical follow up (PMCF) not at all. The MDR exactly describes respective requirements in several articles and a comprehensive annex. - Post-Market Surveillance
The post-market surveillance, that was barely addressed in MDR, now is regulated in detail. - Placing on the market, conformity assessment procedures
The conformity assessment procedures were changed: There is no longer a procedure comparable to Annex VI of MDD. Additionally, for high risk products (some class IIa and most of class III products) additional procedures must be followed and the notified bodies must consult expert groups/panels (scrutiny). - Classification
The classification rules now take active implantable devices, nano-materials, and substances introduced into the body into account. A new classification rule 11 specifically addresses software – major reclassifications are expected. - Common specifications
The EU commission entitles itself to introduce “common specifications” (CS) if harmonized standards are lacking or insufficient. So far there is no standard harmonized nor any CS published.
EU Regulations versus EU Directives
It is also important to understand, that EU regulations are effective in all EU countries with date of publication. In contrast, the EU directives must be implemented in national law first.
There will be still national laws e.g. to define fines. However, the essential requirements manufacturers and importers must comply with are laid out in the MDR.
Switzerland, a non-EU country, stays part of the EU-system.
Main Differences to US / FDA Regulations
The requirements of the new medical device regulations are much closer to the FDA requirements. Nevertheless, manufacturers that already have a FDA clearance must consider:
- A successfully passed conformity assessment procedure is a prerequisite.
- Apart from class I products this means an involvement of notified bodies and a quality management system compliant with ISO 13485.
- Manufacturers must prove conformity typically by applying harmonized standards such as IEC 62304, IEC 62366 and ISO 14971. Fortunately, these standards are very comparable to FDA requirements as expressed in guidance documents.
- Obviously, the medical device reporting is different. This does not only apply to form and addressees.
- The EU has dedicated requirements to “economic operators” such as importers, distributors, EU representatives and to the “person responsible for regulatory requirements”.
- The requirements with respect to clinical evaluation and post-market clinical follow-up are different (partially higher).
Medical Device Regulation (MDR) at a glance
The Medical Device Regulation MDR consists of 10 chapters that group in total 123 articles. MDD consist of 23 articles.
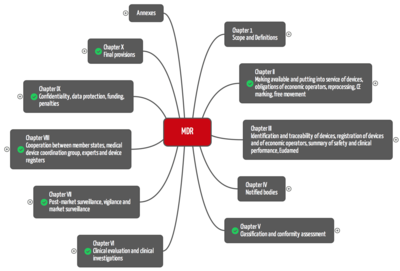
For manufacturers, the most relevant chapters are:
- Chapter II: Making available and putting into service of devices, obligations of economic operators, reprocessing, CE marking, free movement
- Chapter V: Classification and conformity assessment
- Chapter VI: Clinical evaluation and clinical investigations
- Chapter VII Post-market surveillance, vigilance and market surveillance
Additionally, the MDR has 22 annexes:
- I General safety and performance requirements
- II Technical documentation
- III Technical documentation on post-market surveillance
- IV EU Declaration of conformity
- V CE marking of conformity
- VI Information to be submitted upon the registration …
- VII Requirements to be met by notified bodies
- VIII Classification rules
- IX Conformity assessment based on a quality management system and assessment of the technical documentation
- X Conformity assessment based on type examination
- XI Conformity assessment based on product conformity verification
- XII Certificates issued by a notified body
- XIII Procedure for custom-made devices
- XIV Clinical evaluation and post-market clinical follow-up
- XV Clinical investigations
- XVI List of groups of products without an intended medical purpose referred to in Article 1(2)
- XVII Correlation table
MDR: Time line
MDR was published in February 2017. As of May 25th, 2017, MDR "entered into force" and will apply three years later.
Criticism
The MDR increase the burdens especially for small businesses. It will cause bureaucracy, but it is doubtable that the ultimate goal – improving patient safety – will be achieved. Already the MDD and accompanying MEDDEV documents gave enough guidance. Insufficient products were not the result of an insufficient regulation, but of an insufficient application and enforcement of regulations.
Classification rule 11 is one example of how the regulation will cause trouble to medical app start-ups.
MDR will kill innovations. Unfortunately, nobody counts the deaths caused by products that never were invented or marketed since the Medical Device Regulation was preventing innovative companies to come-up new products.

Author:
Prof. Dr. Christian Johner

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.