
Author:
Dr. Bettina Martin
The Clinical Evaluation Plan
Including a free download of the chapter structure for the Clinical Evaluation Plan
The Clinical Evaluation Plan is one of the most complex documents in the technical documentation. Alongside the Clinical Evaluation Report, the Clinical Evaluation Plan (CEP) is the document most frequently criticized by notified bodies.
Even experienced clinical experts typically need 60 working hours to write the document, which is often more than 50 pages long, in such a way that it is accepted by notified bodies.
This article and the chapter structure for free download will help,
- to create the Clinical Evaluation Plan quickly and in compliance with the law,
- avoid the most common errors and thus time-consuming rework,
- to plan the entire development project reliably, and
- bring the device to market without delays and unnecessary hassle.
1. The Clinical Evaluation Plan: Answers to the most important questions
1.1 What is a Clinical Evaluation Plan?
In the Clinical Evaluation Plan, medical device manufacturers determine their strategy for the data and methods they want to use to demonstrate their devices' safety, performance, and benefit and later document them in the Clinical Evaluation Report.
1.2 Is there an obligation to compile a Clinical Evaluation Plan?
Yes, this obligation exists. Article 61 of the MDR obliges all medical device manufacturers to conduct a clinical evaluation. This clinical evaluation must meet the requirements of Annex XIV Part A. It states:
To plan, continuously conduct and document a clinical evaluation, manufacturers shall: (a) establish and update a clinical evaluation plan, which shall include at least […]
MDR, Annex XIV, Part A, Section 1
Consequently, authorities and notified bodies demand this Clinical Evaluation Plan.
1.3 What is the purpose of the Clinical Evaluation Plan?
Manufacturers should create the plan for the following reasons:
- They can avoid regulatory hassles that regularly lead to costly rework and delays placing devices on the market.
- Manufacturers can thereby, at an early stage,
- notice that their planned device does not comply with the state of the art,
- take countermeasures, e.g., by adapting the intended purpose, and
- avoid unnecessary development costs.
- The plan provides clarity about the duration and costs of the development project. For example, during this planning stage, it is decided whether a clinical investigation is necessary.
1.4 Who should write the Clinical Evaluation Plan?
Clinical or medical affairs managers or clinical evaluation specialists have the necessary competencies. These are experts who are proficient in scientific methods and medical writing.
Manufacturers should turn to external service providers such as the Johner Institute if this expertise is lacking.
Attention
Typical product managers usually do not have these competencies. Even training as a doctor is not sufficient to meet the competence requirements.
2. Contents of the Clinical Evaluation Plan
The MDR and the MDCG documents specify the contents of the Clinical Evaluation Plan. Annex XVI Part A of the MDR lists eight points (see Fig. 1).
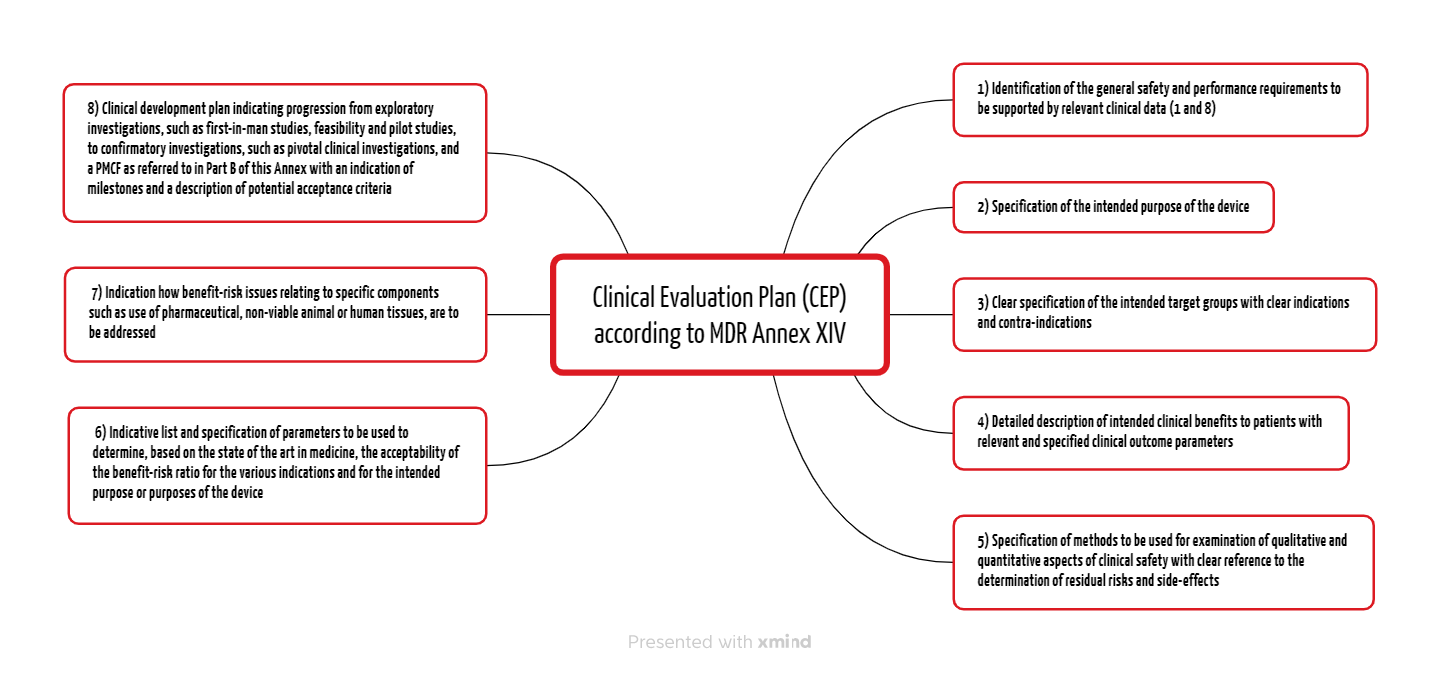
2.1 General safety and performance requirements
Manufacturers must already outline the general safety and performance requirements in the Clinical Evaluation Plan (CEP), which they must demonstrate with relevant clinical data depending on the device and risk. In doing so, they fulfill requirements 1 and 8 from Annex I of the MDR.
2.2 Device
Typically, investigators of notified bodies expect the following information about the device:
2.2.1 Device description
The description of the device shall include:
- a general description of the main functional elements: its parts/components (including software, where applicable), its formulation, its composition, its functioning, and, where applicable, its qualitative and quantitative composition
- the functional principles of the device and its mode of action; an explanation of any novel characteristics
- images or other relevant information, such as diagrams, if necessary, to understand the use of the device
2.2.2 Device classification
Not only the class, but also the applicable classification rules and indents should be stated here.
2.2.3 Device configurations/variants
Manufacturers should also describe the device configurations and variants:
- sizes, differences in design features, different configurations, etc.
- if possible, pictures of these configurations and variants
- description of the history and/or changes since the last evaluation
- reasons for the differences in the design variants
2.2.4 Accesories or compatible devices
- all accessories or compatible devices
- in the case of a system/procedure package, its "components"
- If the use of accessories or compatible devices has an impact on the clinical safety or performance or the scope or validity of the clinical evaluation, this should be stated here.
2.2.5 Intended purpose
- the intended patient population and the medical conditions being diagnosed, treated, and/or monitored
- indications and contraindications
- a detailed description of the intended clinical benefit for patients with relevant and specific outcome parameters for the clinical output (This point is essential to determine the clinical approval strategy for the device. However, manufacturers must already be familiar with the state of the art (see chapter 2.3).)
Note
These elements should be taken verbatim from the intended purpose document or the device description and be identical in all technical documentation documents (e.g., also in the instructions for use). The notified bodies pay close attention to this.
Companies that work with the Johner Institute's digital approval platform do not need to follow this advice. The software automatically adapts these elements and avoids redundant and potentially inconsistent information.
2.3 State of the art
Manufacturers are obliged to determine the state of the art of their device in order to choose the right clinical strategy. A systematic literature search is required to evaluate their device compared to the state of the art.
Tip
This is why the Johner Institute's clinical experts evaluate this state of the art right at the start of development projects.
The state of the art also helps to decide whether clinical data is required for the clinical evaluation of the medical device (if necessary through a clinical investigation) or whether performance data is sufficient.
Further information
The article on literature searches helps with a targeted search and the precise formulation of a legally compliant clinical evaluation.
2.4 Clinical benefit with clinical parameters and acceptance criteria
This allows manufacturers to determine clinically relevant parameters with acceptance criteria in order to demonstrate the clinical benefit. It is helpful to familiarize yourself with the state of medical knowledge and to analyze the risks and side effects that may arise.
Example
A cold pack is intended to relieve pain. The manufacturer determines the achievable skin temperature as a parameter and values below 13 °C as criteria for acceptability.
These preliminary considerations are helpful to
- better understand the complexity of the project,
- support the project team with targeted information,
- weigh up whether certain claims can be made at all,
- decide which risk-minimizing measures need to be taken,
- to determine clinical endpoints of clinical investigations if required.
In the case of existing products, it is necessary to review whether the state of the art has changed and whether the intended purpose and benefits can still be maintained.
Caution
In this step, it may turn out that clinical parameters do not make sense or do not exist and that safety, performance, and benefit can be better demonstrated with technical parameters.
The parameters and the acceptance criteria must correspond to the state of the art.
Further information
In the article on clinical endpoints, you can find out why they are so relevant and how to demonstrate them in clinical investigations.
2.5 Data route and clinical strategy
2.5.1 The alternatives
Once it is clear what needs to be demonstrated, it is important to determine how to demonstrate that the device complies with the state of the art.
There are three main strategies for providing this evidence in clinical evaluation (see Fig. 2):
- First, there is a distinction between clinical data and performance data (3), then
- a distinction between how the clinical data is collected, namely with the company's own device (1)
- or an equivalent product (2).
In the case of clinical data on the company's own device, a distinction can also be made as to whether a clinical investigation is necessary or not. The latter applies if there is sufficient clinical data from PMS and PMCF activities.
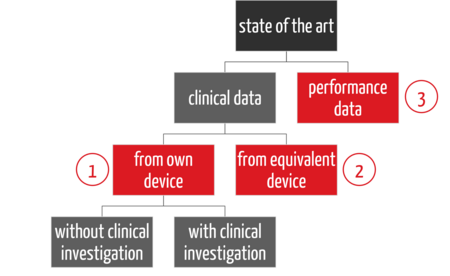
The strategy of providing evidence only via performance data (3), also known as the "data route," is already defined in the Clinical Evaluation Plan. Many notified bodies also require an Appraisal Plan. This describes how which type of data is evaluated.
Example
If you are considering the equivalence route, it is advisable to carry out the equivalence comparison already in the CEP and to describe in the Appraisal Plan which data and criteria you consider a device sufficiently equivalent.
Further information
The article on equivalence describes in detail how to prove equivalence.
Manufacturers are not free to choose the "data route." Several regulatory requirements must be considered (see Tab. 1).
2.5.2 The regulatory requirements
MDR | Data route of the clinical evaluation |
Annex VII, Chapter III | Is the medical device an implantable medical device or a class III medical device? |
Article 61, 6b | Are they sutures, staples, fillings, braces, dental crowns, screws, wedges, dental or bone plates, wires, pins, clamps or connectors? |
MDCG-2020-6 | Is the medical device an existing one under the MDD/AIMDD (= existing device)? |
| Was the device in question designed by modifying a device already placed on the market by the same manufacturer? |
Annex XIV, Part A, 3 | Is there a medical device whose technical, biological and clinical characteristics are similar to the evaluating medical device in such a way that there is no clinically meaningful difference in the safety and clinical performance of the devices? |
Article 2, 48 | Is there clinical data on the medical device from the following sources: (1) clinical investigation(s) of the device in question, (2) reports of other clinical experience with the device in question published in peer-reviewed scientific literature, (3) clinically relevant data from post-market surveillance, in particular from post-market clinical follow-up? |
Article 2, 51 | Does the medical device have clinical endpoints and does it have clinical claims? |
Article 61, 10 | Is the demonstration of compliance with essential safety and performance requirements based on clinical data considered inappropriate? |
Tab. 1: Selected data route for the detection of GSPR 1 and 8
Tip
Do not determine the clinical strategy or data route in the Clinical Evaluation Report, but already in the Clinical Evaluation Plan.
2.6 Methods for investigating clinical safety and performance
Manufacturers must explain how the evidence is to be provided and specify the methods. Possible methods include
- usability tests
- impact and drop tests
- testing of electromagnetic safety
- material characterization
- software integration tests
- penetration tests
- animal testing
- endurance tests
- clinical investigations
- PMS data
- PMCF data
Manufacturers should specify the standards used.
2.7 Clinical evidence
Article 61 of the MDR obliges manufacturers to specify the quality and scope of the data. It calls this "clinical evidence".
Definition “clinical evidence“
clinical data and clinical evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer;
The necessary clinical evidence depends on the device, its risk and the state of the art.
Example
A study with a small patient population may be appropriate if it is a niche device. However, the number of cases may not be sufficient for another device, depending on the risk class.
Manufacturers should also determine this clinical evidence in the Clinical Evaluation Plan.
Note
The clinical evidence, the selected data route, and the optional description of a clinical investigation are part of the so-called clinical development plan.
2.8 Appraisal Plan
In addition, MEDDEV 2.7.1 rev.4 from 2016 requires an "Appraisal Plan" in section 9.2.
This plan should not only take into account the literature assessment. Other data should also be systematically evaluated, for example:
- test reports
- PMS data
- instructions for use
- marketing materials
3. Clinical Evaluation Plan: The chapter structure
Neither the MDR (Annex XIV) requirements nor the MDCG 2020-6 for existing products (Annex II) enforce a chapter structure. However, it should be chosen in such a way that it leads the reader from the general to the specific.
Download
You can download a proposal for such a chapter structure here.
4. The Clinical Evaluation Plan in the development process
The Clinical Evaluation Plan is a document that manufacturers should prepare very early in the development process because, as part of this plan, they determine the state of the art. This "state of the art" must be known and considered in other documents and content (see Tab. 2).
Document, Contents | Comments |
Intended purpose | The intended purpose must correspond to the state of the art. Conversely, the claims that must be proven also follow from the intended purpose. The Clinical Evaluation Plan determines how this evidence is provided. |
Clinically relevant parameters with acceptance criteria | These parameters and acceptance criteria usually result in product requirements. |
Preliminary risk analysis | This can result in risk-minimizing measures or requirements for the device. |
Necessity and design of the clinical investigation | For example, the clinical evaluation must review whether the above-mentioned acceptance criteria are met. The endpoints of these studies must correspond to the state of the art. |
Tab. 2: Documents for which the state of the art is a requirement
The information influences each other (see Fig. 3).
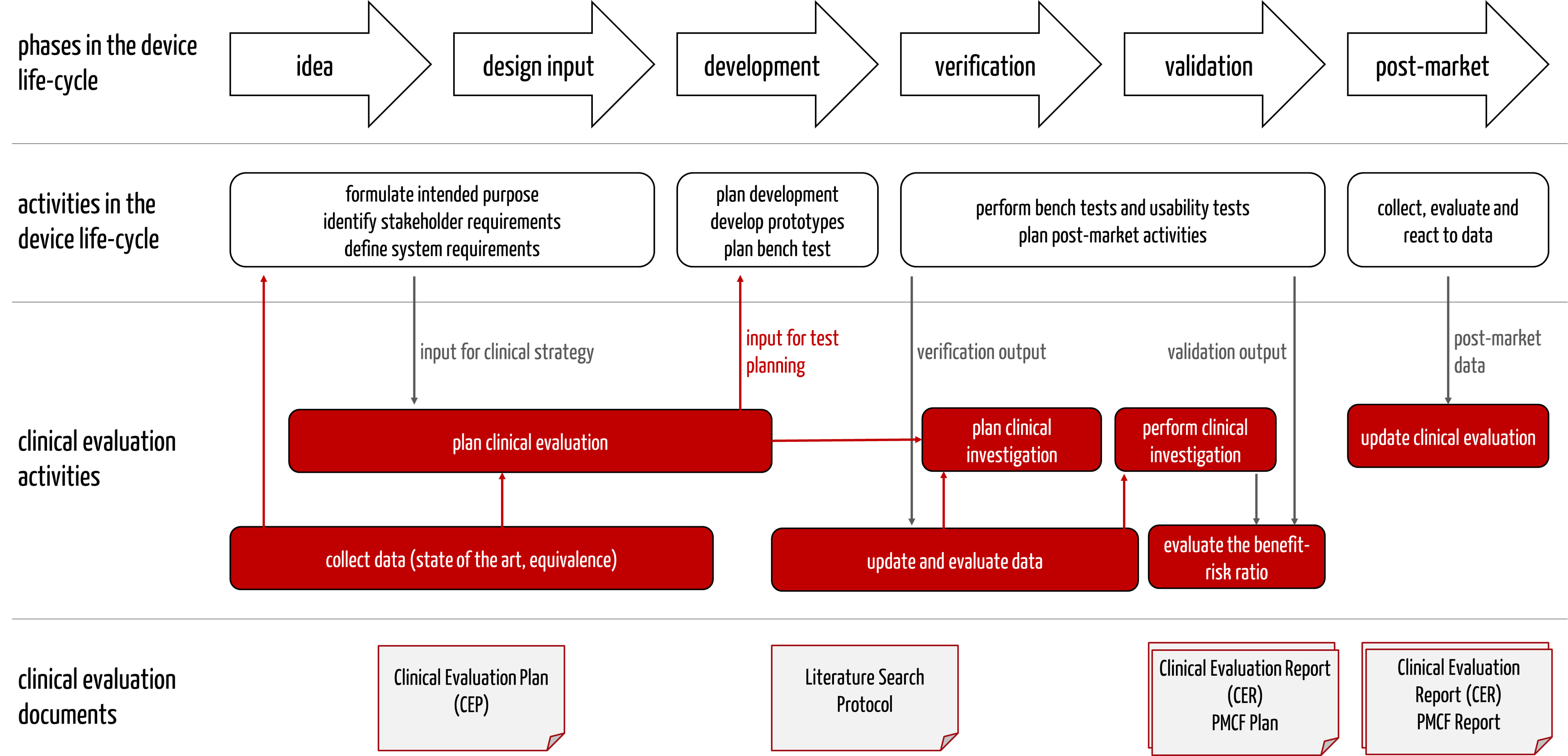
Companies should pay attention to this and design the processes so that the clinical affairs experts are involved in formulating the intended purpose from the outset. Otherwise, there is a risk of setbacks and unnecessary effort and delays.
5. Recognize and correct typical errors
Error | Correction |
The manufacturer does not explain his device but only references other documents. | Both the CEP and CER contain this information (unfortunately) redundantly, e.g., intended purpose, indications, and contraindications are identical to the other documents. |
The clinical benefit is missing. | The manufacturer specifies clinical claims and quantifies them with parameters (see above). |
The manufacturer does not specify any acceptance criteria for the parameters. | The manufacturer determines one or more acceptance criteria for each of these parameters. |
The appraisal plan is missing completely or not detailed enough. | The manufacturer should clarify with the notified bodies whether they require an "Appraisal Plan" and, if so, in what form. |
The clinical strategy is not clear. | The manufacturer describes exactly how he will demonstrate safety, performance, and benefit. Among other things, the manufacturer shall determine whether this is based on clinical data and whether this data originates from its own device or an equivalent product. |
The plan is not adapted to existing products (see MDCG-2020-6). | The Clinical Evaluation Plan also needs to be updated. For example, the clinical strategy for existing products can be covered with PMS and PMCF data and sufficient clinical evidence. |
The manufacturer starts planning the clinical evaluation after the final formulation of the intended purpose or even during the verification of the device. | The clinical evaluator determines the clinical strategy while formulating the intended purpose. If this is not done, the intended purpose may contain clinical claims that would later have to be proven with a clinical investigation, although a different route could perhaps have been taken via performance data. |
The manufacturer communicates with its notified bodies only after the clinical evaluation has been written. | For new products with innovative aspects in particular, it is advisable to consult with the notified bodies in advance to ensure that they are on board with the chosen clinical strategy. Although notified bodies are not permitted to provide advice, they are allowed to provide information on whether the clinical strategy is appropriate. These queries to the notified body's clinical reviewer are also recommended before correcting any defects that have been criticized. |
Tab. 3: Typical errors and how to correct them
6. Summary and conclusion
6.1 A challenging task
Creating a Clinical Evaluation Plan is one of the most challenging tasks:
- The competence requirements are very high. This applies to scientific competence as well as the necessary knowledge of the device and the medical context.
- The document is very extensive and "content-heavy." Compiling the data is very time-consuming. Even experienced clinical affairs experts need one to two weeks to do this.
- The preparation requires close and intact interaction with almost all areas of the company and has an impact on the entire product life cycle.
- There seems to be no consensus among notified bodies regarding the requirements for this plan. The requirements differ not only between notified bodies but also within them. This also leads to regular deviation reports.
Even if the task is difficult and time-consuming, manufacturers should not put it off. After all, by drawing up a valid and precise clinical evaluation plan, they have already made groundbreaking decisions.
6.2 Groundbreaking specifications
By writing the Clinical Evaluation Plan, the manufacturers ...
- ensured that the device to be developed will comply with the state of the art.
- identified risks and derived measures and product requirements.
- defined the necessary methods for product testing.
- clarified whether a clinical investigation is necessary (an essential prerequisite for project planning).
- laid the foundations for a compliant clinical evaluation report and, thus, the approval of the device.
6.3 The nuts and bolts of clinical evaluation
The Clinical Evaluation Plan is one of the first documents manufacturers should start with. It has an impact on other documents, such as the intended purpose and the design input.
Conversely, the Clinical Evaluation Report is one of the last documents that the manufacturer finalizes because it relates to almost all other technical documentation documents.
In other words, the Clinical Evaluation Plan is the alpha, and the Clinical Evaluation Report is the omega not only of the clinical evaluation but of the entire product development.
Both documents must be updated on an ongoing basis.
Support from the Johner Institute
The Johner Institute's clinical experts will help you prepare your Clinical Evaluation Plan in the shortest possible time so that it will be approved by your notified bodies. You will then have a solid basis, including for your Clinical Evaluation Report.
The assessment plan also implicitly provides you with your clinical strategy.
- You ensure that your planned device corresponds to the state of the art and may be marketed.
- You know the risks of your device and can define measures at an early stage, which is faster and cheaper than doing it later.
- You know what evidence you need to provide and by what methods. This helps you to estimate the effort involved and create a realistic project plan.
- You can commission pre-clinical tests or a clinical investigation in good time to keep to the schedule and save yourself stress.
Minimize your (not only) regulatory risks and benefit from the help of the Johner Institute's clinical experts. Get in touch now.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.