Conformity Assessment Procedures according to MDR
Medical device manufacturers have to follow conformity assessment procedures before placing products on the market. With these conformity assessment procedures manufacturers must prove compliance of the products with the essential requirements laid down in the Medical Device Directive (MDD) respectively Medical Device Regulation (MDR).
Definition: Conformity assessment
‘conformity assessment’ means the process demonstrating whether the requirements of this Regulation relating to a device have been fulfilled“
Source: MDR
Conformity assessment procedures: It all depends on the class
Both the MDD as well as the MDR offer a variety of conformity assessment procedures. However, which of these procedures may be followed depends on the class of the medical device.
More Information
Read more about the classification of medical devices.
For medical devices that fall into class I, the conformity assessment does not involve a notified body. For all other classes (I*, IIa, IIb, III) a notified body must be involved. The number of the notified body appears on the CE-mark.
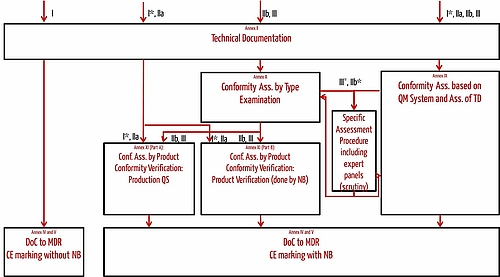
Overview on Conformity Assessment Procedures
For medical devices that fall in class I, manufacturers declare the conformity and affix the CE-mark as described in Annexes IV and V.
For products that fall in higher classes either a full quality management system (compliant with ISO 13485 respectively Annex IX) is required or the manufacturers have to follow Annex XI.
Annex IX offers two alternatives:
- Part A describes the demands on a quality management system for the production.
- Part B describes how a notified body has to verify the conformity of each single instance.
For class IIb and III devices also a type examination is necessary as described in Annex X (if not Annex IX is followed). This type examination means that the manufacturer develops a medical product. Before producing and selling the product the notified body verifies the conformity of this device with the "general safety and performance requirements".
The Annexes of MDD and MDR roughly correspond like follows:
| MDR | MDD |
|---|---|
| Annex IX | Annex II |
| Annex X | Annex III |
| Annex II | Annex VII |
| Annex XI part A | Annex V |
| Annex XI part B | Annex IV |
| -- | Annex VI |
Special Procedures
For high-risk products special procedures have to be applied. These special procedures include a scrutiny by an expert panel.
Example for high-risk products are:
- active implantable products
- devices containing medicinal products
- devices containing tissue from human or animal origin
- "devices that are composed of substances or of combinations of substances that are intended to be introduced into the human body"
The expert panel has to judge whether the clinical evaluation report is sufficient to provide confidence in the safety and performance of the device and whether an additional scientific assessment report (by the expert panel) is required.

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.