Regulation (EU) 2017/746: In vitro Diagnostic Medical Device Regulation (IVDR)
February 2024
The European In Vitro Diagnostic Medical Device Regulation (IVDR) must be followed by manufacturers who wish to place in vitro diagnostic medical devices on the market in the EU.
The Regulation (EU) 2017/746 In vitro Diagnostic Medical Device Regulation (IVDR) regulates the entire life cycle of in vitro diagnostic medical devices (IVDs) in the European market. The IVDR came into force on May 25, 2017 at the same time as Regulation (EU) 2017/745: Medical Device Regulation (MDR), which regulates all other medical devices and their accessories. The IVDR has been valid since May 26, 2022. A timeline and explanations of the transitional provisions can be found in this article.
In this article, we would like to give you an overview of the IVDR, the relevant stakeholders, its structure, the requirements, as well as background information on the legal act. We have provided links to other articles from the Johner Institute on the respective topics to help you familiarize yourself with this sometimes difficult topic.
1. Who does IVDR concern?
1.1 Scope
The IVDR is responsible for the entire market for IVDs in the EU, from development and market surveillance through to use. In particular, the IVDR regulates the requirements that IVDs must fulfill to be placed and operated on the European market.
Definition in vitro diagnostic
An in vitro diagnostic is defined as a medical device that is intended by the manufacturer to examine samples derived from the human body and provide relevant diagnostic information. This information may include, but is not limited to, physiological or pathological conditions, predispositions to disease, anticipated effect of treatments, and therapy monitoring.
Other products such as reagents, calibrators, control materials, instruments, and specimen receptables are also considered in vitro diagnostics under the IVDR.
1.2 Affected organizations
- Thus, the main addressee of this Regulation are the manufacturers of IVDs.
- Other economic operators who must comply with the requirements of the IVDR are distributors, importers, manufacturers from third countries, and their authorized representatives.
- In addition to the economic operators, the IVDR also imposes requirements on the competent national authorities and the notified bodies, which are included in the conformity assessment of a significantly wider range of IVD products in comparison to the old IVDD.
- The third sub-group, which is held responsible by the IVDR, are health institutions such as hospitals and clinics that operate IVDs or laboratories that act as manufacturers of self-developed in-house IVDs.
1.3 Further tips
IVDR Tips
... for beginners
If you're new to the topic, download the free starter kit. It provides you with an overview of the regulations, shows you the steps to conformity of your medical device, and contains the IVDR checklist for download.
... for advanced readers
Work with the consolidated version of the IVDR in English or German. This summarizes all changes to the IVDR, including the transitional provisions extended in March 2023. Internal links make it easier for you to navigate the more than 170-page regulation.
Go deeper into the details with the articles linked in the IVD article section.
... for manufacturers of IVDs according to Directive 98/79/EC (IVDD)
Manufacturers who have already placed IVDs on the market in accordance with the IVDD should read the article on the differences between IVDR and IVDD. The chapters of the IVDR have been completely restructured compared to the old In Vitro Diagnostic Directive (IVDD). In the linked article, you will find information on the changes in IVDR compared to IVDD.
... for manufacturers of medical devices
Manufacturers of medical devices should read the article on the MDR.
... for importers, distributors, hospitals, and other operators, as well as laboratories and RUO manufacturers
Please refer to the related linked articles in accordance with Chapter 3.2.
2. Structure of the IVDR
The IVDR comprises 113 articles, which are divided into ten chapters. Figure 1 shows the structure of the IVDR with its chapter topics. We have summarized the contents of the ten chapters for you in Table 1.
The 15 annexes of the IVDR are not to be neglected. They specify many of the requirements described in the articles of the IVDR. For example, Annex I contains the General Safety and Performance Requirements that apply to all IVDs and must therefore be met. We have summarized the contents of the 15 annexes in Table 2.
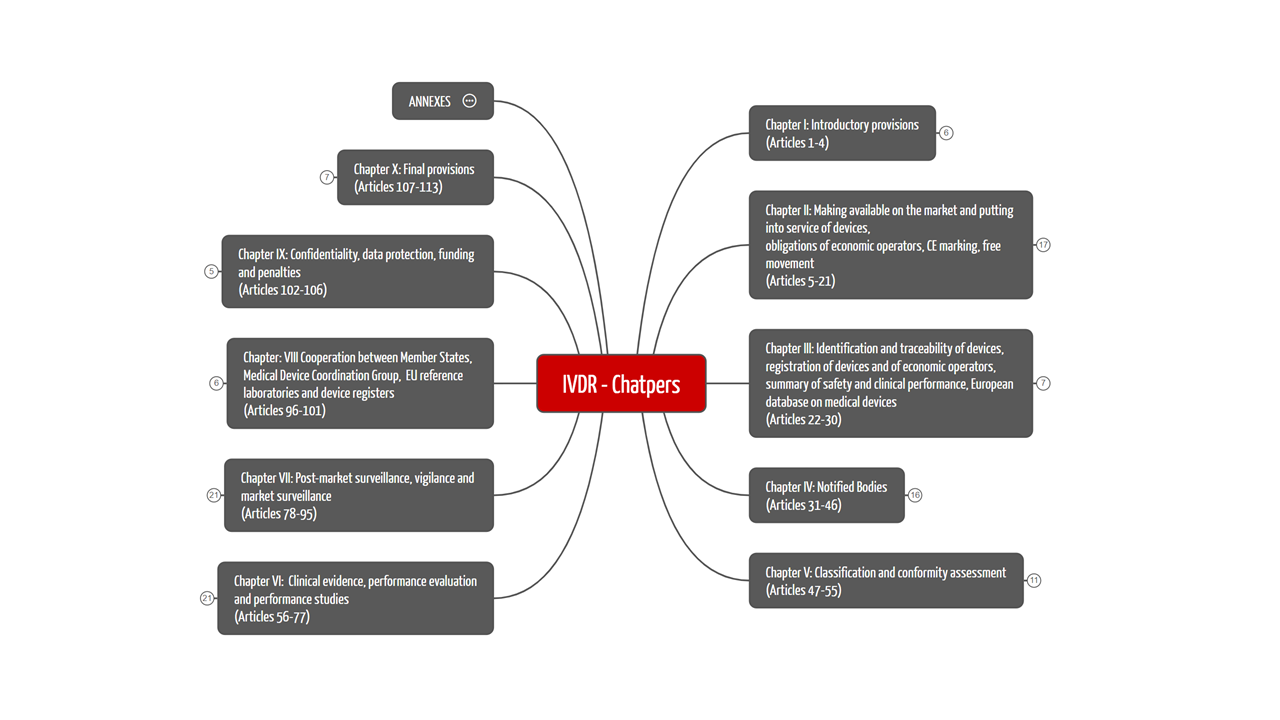
The mind map with annexes is part of the free starter kit, which gives you a quick overview of the more than 170-page IVDR.
2.1 The ten chapters of the IVDR
In Table 1 we have provided you with a clear overview of the chapter structure and its contents.
| Chapter Number (With links to IVDR) | Title / Content / Requirements (With links to articles) |
| I | Scope and Definitions |
| II | Requirements for Manufacturers, Distributors, Importers, and Member States. Many references to articles in other chapters and to the annexes. |
| Traceability of Products, esp. UDI Requirements | |
| IV | Requirements for Notified Bodies |
| V | Classification, Authorization, and Conformity Assessment Procedure |
| VI | Performance Evaluation and Performance Studies |
| VII | Post-Market Surveillance, Reporting |
| VIII | Cooperation between Member States, MDCGs, and other Experts |
| IX | Confidentiality, Privacy, Penalties |
| X | Transition Provisions and more |
Table 1: The chapter structure of the IVDR
2.2 The 15 Annexes to the IVDR
The IVDR has 15 annexes that address various important topics related to the conformity of IVD devices. Figure 2 and table 2 show the contents of the annexes.
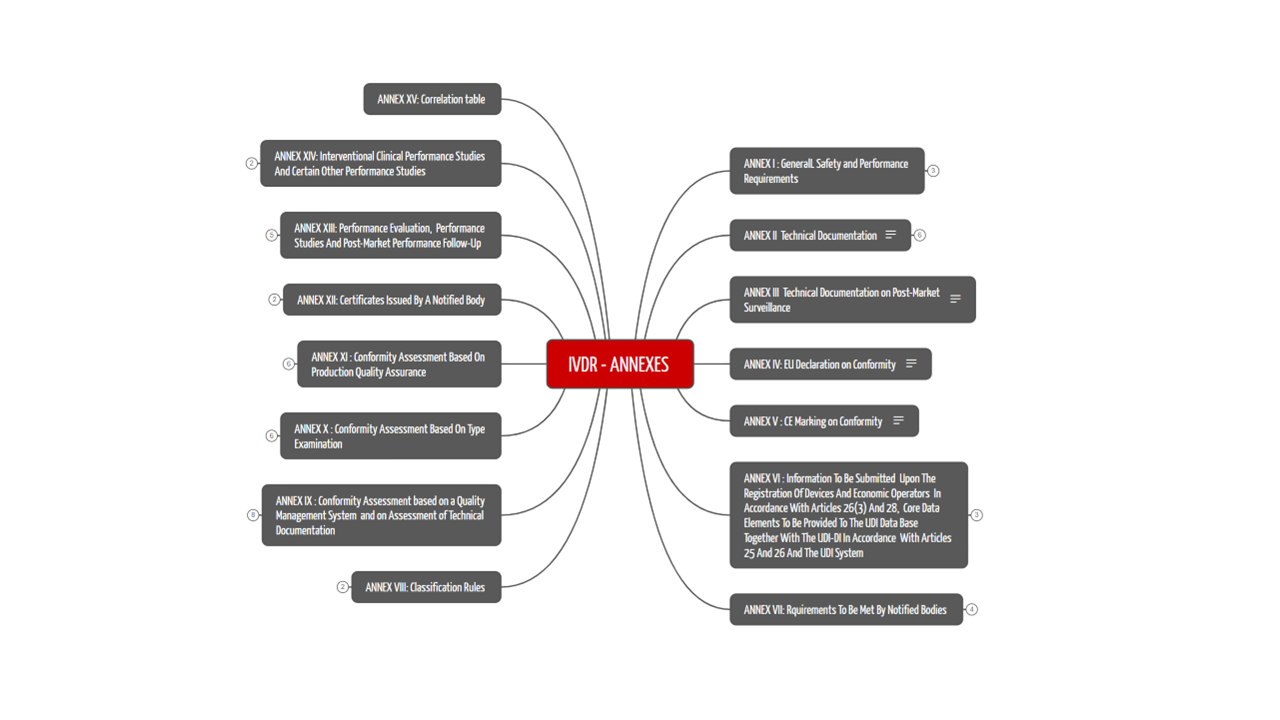
Annex number | Title / Content / Requirements |
Technical Documentation for Post-Market Surveillance | |
Requirements for Notified Bodies | |
Conformity Assessment Procedure: complete QM System | |
Certificates issued by Notified Bodies | |
Performance Evaluation, Performance Studies, and Post-Market Performance Follow-Up | |
Interventional Clinical Performance Studies | |
Correlation Table |
Table 2: The Annexes of the IVDR
Information on the technical documentation, which manufacturers use to prove the conformity of their products, can be found directly in the first annexes. In addition, topics such as registration in EUDAMED, UDI, classification rules, and declaration of conformity are explained in the IVDR annexes. Performance evaluation and clinical performance studies are also covered in the annexes of the IVDR.
3. Overview of the requirements of the IVDR
Now that the design and structure of the IVDR are known, we will look at the requirements for manufacturers (3.1) and the requirements for other economic actors such as distributors, importers, and laboratories (3.2).
3.1 Requirements for the manufacturers themselves and their IVDs
Table 3 shows the requirements for IVD manufacturers. If the manufacturer seeks IVDR compliance for one of its IVDs, he must establish a quality management system and maintain it throughout the entire life cycle of the products. ISO 13485 is the harmonized standard to be regarded here.
3.1.1 Requirements for the manufacturers
Requirement of the IVDR | Explanation |
All manufacturers need a QM system, among other things for the development, production, and monitoring of products on the market. With the exception of class A devices, the QM system is audited by the notified bodies as part of the conformity assessment procedure. For class A sterile devices, the involvement of a notified body is limited to "the aspects of manufacture related to achieving and maintaining sterile condition" (according to IVDR, Art. 48 (10) and Annex XII, Chapter I, No. 7.)". | |
Manufacturers apply a risk management system to ensure that the company-specific risk policy is implemented and adhered to throughout the life cycle of the individual products. The risk management file, on the other hand, represents the product-specific risk-benefit assessment. | |
Manufacturers are obliged to employ a person who is responsible for regulatory compliance (according to IVDR, Art. 15; also called PRRC) and ensures, for example, that the technical documentation is prepared in accordance with their own QM system. EU authorised representatives must also have access to a PRRC. |
Table 3: Requirements for manufacturers, their systems, and roles
3.1.2 Product-specific requirements
In addition to the cross-product requirements, there are also requirements for each individual product. These product-specific requirements result from the intended purpose of the respective IVD. This, in turn, results in the qualification as an IVD, the classification into one of the four risk classes, the requirements for performance evaluation, product-specific risk management, and labeling. Further verification and validation activities, such as usability or stability verifications, as well as the development file, are also part of technical documentation. Post-market surveillance and vigilance are also product-specific tasks that manufacturers must fulfill according to the IVDR.
Another new obligation for manufacturers is the assigning of a UDI for each IVD and its registration in EUDAMED. This is intended to facilitate traceability and post-market monitoring for all stakeholders. In summary, the technical documentation of an IVD according to Annexes II and III serves to prove that all valid general safety and performance requirements (Annex I) are met. Table 4 provides an overview of these product-specific requirements.
Requirement of the IVDR | Explanation |
Classification of IVDs and Classification of Software IVDs | For each IVD, the manufacturer must determine the risk class, which is largely determined by the intended purpose. |
General Safety and Performance Requirements & Technical Documentation | The technical documentation must comply with the requirements of Annex II and provide all evidence that the general requirements of Annex I are met. For this evidence, manufacturers are required to apply harmonized standards and the common specifications. |
Risk Management | For each IVD product, the manufacturer must identify, assess, and manage product-specific risks. In a continuous process, the manufacturer must constantly assess the risk-benefit ratio. |
As part of the performance evaluation, the manufacturer must continuously check whether the safety and performance of the IVD are given. If the clinical data is not sufficient, a (clinical) performance study is necessary. | |
All IVDs must be given a unique identification, the UDI. The products must be registered in EUDAMED. | |
Labeling | The IVDR specifies requirements for the instructions for use, other accompanying materials, and other labelling such as imprints and packaging. |
Conformity Assessment and Approval | Depending on the risk class of the product, the manufacturer may go through a conformity assessment procedure. In doing so, the manufacturer must involve a notified body, except in the case of Class A devices. If successful, he must create a declaration of conformity and affix the CE mark. |
Manufacturers are obliged to monitor their IVDs over their entire service life on the market, to continuously collect data on this and, if necessary, to react to it. If they identify risks, they must inform the authorities. |
Table 4: Requirements of the IVDR for the devices
3.1.3 Note on fulfilling the requirements
Caution!
It is not only the IVDR that places demand on IVDs and their manufacturers. Many other laws and standards must be regarded. While the IVDR is legally binding, standards and policy documents are not. Nevertheless, in reality, a manufacturer will have difficulties in proving the conformity of its processes and products if it does not comply with the harmonized standards applicable to its product.
3.2 Requirements for other (economic) actors
In addition to IVD manufacturers as addressees of the IVDR, other stakeholders are affected. Table 5 shows a list of other (economic) actors within the scope of the IVDR.
Actors | Explanation |
IVD distributors are the only economic operators that are not required to register with EUDAMED under the IVDR. However, they are not exempt from obligations, such as forwarding and reporting incidents and feedback from users. | |
Importers of IVDs from third countries
| Importers who place products from a third-party country on the Union market are subject to additional obligations under the IVDR. Unlike distributors, importers must register with the EUDAMED. |
Manufacturers outside the EU member states must appoint an EU authorized representative to serve as a point of contact for national authorities. The minimum tasks to be performed by EU authorized representatives are described in Article 11 of the IVDR. In addition to registering in EUDAMED, EU authorized representatives must have access to a Person Responsible for Regulatory Compliance (PRRC) as described in Article 15. | |
Hospitals and Health Institutions
| Some of the articles of the IVDR are also aimed at health institutions. In addition to the IVDR, there are national laws to be complied with. |
Laboratories that act as Manufacturers of in-house IVDs
| Laboratories that offer IVDs developed in-house produce so-called in-house IVDs. For this purpose, general requirements of the IVDR must be met, such as the requirements of Annex I, IVDR. |
Suppliers of RUO Products
| "For Research Use Only" (RUO) devices are not designed for human diagnostics and therefore do not fall within the scope of IVDR. Laboratories that use these products to provide diagnostic tests are in-house IVDs and are therefore regulated by the IVDR. Manufacturers, distributors, and operators who incorrectly apply the RUO label in order to circumvent regulatory hurdles endanger the health of patients and face legal penalties. |
Pharmaceutical Manufacturers in the context of Companion Diagnostics (CDx)
| Manufacturers of companion diagnostics for their drugs are also regulated by the IVDR. The requirements for CDx are higher than for IVDs of the same risk class. |
Table 5: Requirements of the IVDR for other economic actors
4. Background information on the act
4.1 How EU Regulations 2017/175 MDR and 2017/176 IVDR came about
It is still unclear why the EU tackled the new regulation of the medical device and IVD market in 2017. The breast implant scandal is discussed as a trigger event, but this is denied by the actors involved. However, the aim of the regulations is to ensure the safety and health of patients, as well as a barrier-free EU market for medical devices.
4.2 Difference between EU regulations and EU directives
The "old" EU Medical Device Directive, like all EU directives, had to be transposed into national laws and national regulations to become applicable as national law. In Germany, this was realized by the Medizinproduktegesetz (MPG).
The EU regulations (here: MDR, IVDR) have a direct legal character. National laws only supplement these regulations with penal provisions and the determination of the competent authorities.
4.3 Changes to the IVDR compared to the IVDD
This article highlights the changes introduced by the IVDR compared to the old IVDD, the In vitro Diagnostic Device Directive.
5. Conclusion and Summary
The In Vitro Diagnostic Device Regulation (IVDR) is a very comprehensive body of legislation that poses major challenges for all stakeholders (manufacturers, notified bodies, importers, distributors, laboratories).
In contrast to the IVDD, the IVDR has not only significantly increased the regulatory requirements but also expanded the scope of validity. Products such as in-house IVDs (labdeveloped tests, LDT) are now subject to IVDR.
The actors have no choice but to deal intensively with this body of legislation and to meet its requirements. These increased requirements apply to both the "authorization" (pre-market phase) and, explicitly, the post-market phase (post-market surveillance, vigilance).
6. Further articles and links
6.1 Classification
The risk class of an IVD is decisive for the regulatory effort that manufacturers must pay to prove compliance. In this article, you can get an overview of the classification of IVDs according to EU Regulation 2017/746 and you will be instructed on how to classify products in a clever and legally compliant manner.
6.2 Transitional provisions
The EU has postponed the transition periods for the IVDR several times. The rules are so complex that an article on the transitional provisions will help you.
6.3 Special IVDs
In another article by the Johner Institute, so-called sampling kits are discussed in detail. From a regulatory point of view, these sets are close to the MDR's treatment units. In short, several products can be combined into a sampling set if their intended purpose is not changed.
IVDs for self-administration (self-testing) and patient-related tests have been an important part of the healthcare system in Europe, not only since Corona. In this article, we have summarized the regulatory specifics for these IVD products.
6.4 Further regulatory requirements
There are many other laws and standards to be observed, which are presented in the article on the approval of IVDs.
Further explanations and requirements have been published by the Medical Device Coordination Group (MDCG).
Assistance with the implementation of the IVDR
Help from the Johner Institute
Free Offers
Do you have any questions about IVDR and its implementation? You can find answers in our free micro-consulting.
Download the free Starter Kit, which gives you an overview of the regulatory landscape and includes the IVDR checklist in PDF and DOCX format..
Videos and e-learning
Our video trainings show you step-by-step instructions on how to create your technical documentation and your QM system in a lean, fast, and IVDR-compliant way. More than 100 templates and sample documents are available for download. In this way, you create the conditions for your products to be approved and brought to market quickly and safely.
Product testing
The experts at the Johner Institute will help you test your products through:
- Usability Testing
- Penetration Testing
- Test of electrical safety and EMC (e.g., IEC 61010-1)
- Biocompatibility testing
- performance evaluations and performance studies
Consultation
Take advantage of the knowledge and experience of our regulatory affairs experts to:
- determine your regulatory strategy and classify products,
- prepare the technical documentation (TD),
- set up QM systems (QMS), and
- take over the Post-Market Surveillance.
With the IVDR Readiness Check, you can quickly get clarity on how compliant your QM system and technical documentation with the IVDR are. This helps to avoid unnecessary difficulties and delays in audits and reviews of the technical documentation and thus the marketing of the products.
Get in touch now so that we can clarify together how you can quickly and easily meet the regulatory requirements of the IVDR and bring your products safely to market.
Change history (as of May 2021; Remainder deleted)
- 2024-02-25: Chapter numbering revised, subheadings added
- 2024-01-25: Article revised
- 2023-05-03: Article completely rewritten

Author:
Dr. Kai Moritz Eder

Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.